大前庭水管综合征及其相关的基因_SLC26A4
- 格式:pdf
- 大小:116.74 KB
- 文档页数:4



广州地区大前庭水管相关SLC26A4基因热点突变分子流行病学分析目的分析广州地区正常听力人群中SLC26A4基因热点突变IVS7-2A>G和2168A>G发生频率,初步探讨SLC26A4基因热点突变在广州地区人群中的携带率及进行分子诊断的意义。
方法针对2013年1月至2013年9月來我院就诊的6437例正常听力人群均抽取外周静脉血并提取DNA,以博奥生物有限公司提供的晶芯?九项遗传性耳聋基因检测试剂盒(微阵列芯片)对SLC26A4基因对GJB2、SLC26A4、mtDNA12srRNA和GJB3四个耳聋相关基因的9个致聋突变位点进行检测,分析热点突变位点IVS7-2A>G和2168A>G的发生频率。
结果在6437例受检者中,携带SLC26A4基因IVS7-2A>G和2168A>G突变的例数共计107例,总阳性检出率为1.66%。
IVS7-2A>G突变携带者98例,2168A>G突变携带者9例。
结论在6437例正常听力人群中,通过SLC26A4基因热点突变IVS7-2A>G 和2168A>G检测发现广州地区人群该位点的突变携带率为1.66%,低于全国其它地区的所报道的数据。
该调查有助于了解广州地区正常人群中与大前庭水管综合征密切相关的SLC26A4基因热点突变的携带情况,同时也对IVS7-2A>G和2168A>G突变检测阳性患者的婚配、生育具有一定的指导意义。
标签:遗传性耳聋SLC26A4基因突变携带耳聋是临床上最常见的遗传病之一,严重影响患者的社会交流和生活质量。
资料表明,平均每1 000名新生儿中就有1名患有先天性耳聋,其中50%~70%可能与遗传因素相关,基因突变是引起遗传性耳聋的主要因素[1]。
迄今,与NSHL 相关的一些基因已被克隆,遗传性耳聋的病因学研究有了很大进展。
随着基因芯片技术的逐渐成熟,耳聋相关基因芯片已经进入临床应用阶段,为临床耳聋基因的大批量筛查提供了可能条件。


114筮宣匡堂皇王盘查至Q!垒生垒旦箜至鲞箜至塑』望!!!!!卫坐!堕坐鱼:垒匹垫!垒:y!!至:盟!.兰大前庭水管综合征1例家系基因突变分析病例报告季春燕朱海燕(中国人民解放军海军总医院妇产科优生优育中心实验室,北京100048)患儿女,7个月,家长发现其对声音无明显反应,于2013年来本中心就诊。
耳声发射听力检查:左耳2000H z、4000H z可通过,其余均未通过;右耳均未通过。
听觉诱发电位检查:双耳短声反应阈均未引出,波形分化差,重复性差。
双耳容积计算机扫描(V C T)平扫见:双侧前庭导水管扩大,呈喇叭口状,余内耳结构及其他骨质结构均未见异常。
临床诊断为先天性耳聋,大前庭水管综合征(1ar ge ves t i b ul ar aqueduc t syndr om e,L V A S)。
父母听力正常,家族中无同类患者。
基因诊断因该综合征为常染色体隐性遗传病u J,建议该家系进行致病基H SLC26A4的突变分析。
签署知情同意书后,抽取患儿及父母外周静脉血2m l,提取基因组D N A。
首先对患儿进行致病基因21个外显子的扩增,PC R扩增引物及条件参照参考文献[2]。
扩增产物经凝胶电泳回收纯化后,A B I3130测序仪双向测序。
应用D N A st ar 软件对测得序列与标准序列(N C一000007.14)进行比对,分析是否存在突变。
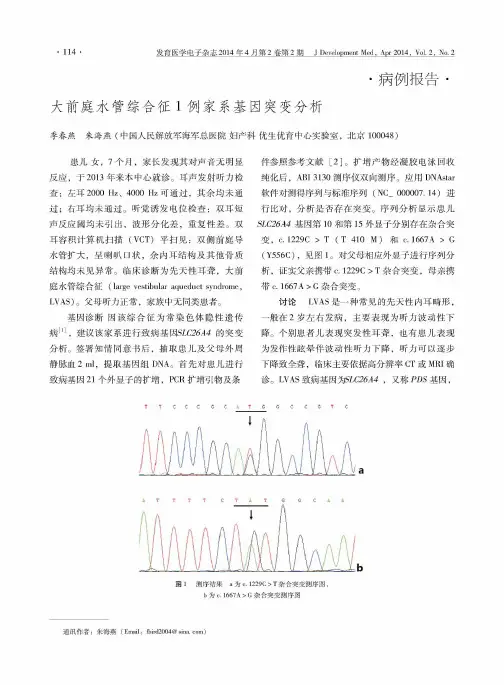
序列分析显示患儿SL C26A4基因第10和第15外显子分别存在杂合突变,c.1229C>T(T410M)和c.1667A>G (Y556C),见图1。
对父母相应外显子进行序列分析,证实父亲携带c.1229C>T杂合突变,母亲携带c.1667A>G杂合突变。
讨论L V A S是一种常见的先天性内耳畸形,一般在2岁左右发病,主要表现为听力波动性下降。
个别患者儿表现突发性耳聋,也有患儿表现为发作性眩晕伴波动性听力下降,听力可以逐步下降致全聋,临床主要依据高分辨率C T或M R I确诊。

大前庭导水管综合征早期诊断中耳聋基因及听力联合筛查的作用【摘要】目的:研究大前庭导水管综合征(LVAS)早期诊断中耳聋基因及听力联合筛查的作用。
方法:回顾性分析200例LVAS患儿在耳聋基因及听力联合筛查后的作用。
结果:听力筛查、SLC26A4基因筛查、耳聋基因与听力联合筛查对LVAS患儿的筛查差异较大(P<0.05)。
结论:耳聋基因及听力联合筛查LVAS,有助于尽早诊断LVAS。
【关键词】大前庭导水管综合征;早期诊断;耳聋基因;听力联合筛查大前庭导水管综合征(LVAS)属于一类常见先天内耳畸形,和SLC26A4基因突变相关,发病率在儿童感音神经性聋中占比较高。
患者发病具有进行性、波动性,会使患者损失感音神经性听力,患儿出生时听力接近正常,多数受感冒、外伤等因素影响,无形中会使其听力降低[1]。
因此,早期诊断对LVAS有良好的预防作用。
当前,新生儿听力筛查广泛开展,影像学检查同样受到听力障碍患者的重视,既往研究已得出听力特点对早期诊断的效用,但众多研究得出就诊年龄主要于幼儿期、学龄前期,临床发现LVAS早期无法就诊[2]。
本研究现对所选的LVAS患儿实施耳聋基因及听力联合筛查,分析效果,现报道如下。
1 资料与方法1.1 临床资料我院2020年1月-12月首诊的LVAS患儿200例作为研究对象,初次听力诊断年龄2月龄-12岁(7.12±2.02),男101例,女99例。
1.2 方法统计全部患儿初次诊断基本特点,包含首诊年龄、听力损失程度等。
按照就诊状况,分析婴儿期未及时就诊的原因,对婴儿期未及时就诊的原因展开单因素分析,分析耳聋基因及听力联合筛查对LVAS的作用。
1.3 检查方法1.3.1 听力检查全部患儿均行声导抗、听性脑干反应(ABR)等。
按较好耳500、1000、2000、4000Hz平均听阈对听力损失程度进行判断,按照WHO标准分级患儿听力损失程度。
ABR反应阈依据美国婴幼儿听力联合委员会所制标准来判断。

大前庭导水管综合征大前庭导水管综合症(LVAS)又称为先天性前庭水管扩大,为一种先天性遗传性疾病,与常染色体隐性遗传有关;是最常见的一种先天性内耳畸形。
研究表明:先天性大前庭水管综合征的发病率为1.5%,占先天性内耳畸形的31.5%。
1791年Carlo Mondini 教授首先观察到了前庭水管扩大这一现象;1978 年,Valvassori 在3700 例听觉或前庭功能障碍患者的颞骨多轨迹断层摄影中,发现50 例患者(1.5%)前庭水管扩大,并将其命名为大前庭水管( large vestibular aqueduct , LVA) ;临床上伴有听力下降等症状者称为大前庭水管综合征。
先天性大前庭水管综合症,可从出生到青春期任何时期发病;典型的临床表现为出生时行听力筛查未通过,于3月内行听力诊断检查发现听力下降,经颞骨CT或内听道MRI影像学检查时确诊。
部分患儿出生时听力正常,但于婴儿期或年幼时才出现听力下降,表现为渐进性、波动性,发病形式可突然或隐匿,常于感冒和外伤时诱发。
在深入了解大前庭导水管综合症这一疾病之前,本期微信我们首先为大家简要介绍一下前庭水管的解剖及基本生理。
1-前庭水管的解剖前庭水管是一个细小的骨性管道,从前庭延伸到颞骨的岩部,包裹着充满了内淋巴液的膜性内淋巴管;作用是将前庭的膜迷路与颞骨内的内淋巴囊相连,正常的前庭水管是维持内淋巴液代谢所必需的前庭导水管是一骨性小管, 呈倒“J ”形,如下图 6 所示。
成人的前庭水管的平均长度约为10 mm , 可分为近侧段和远侧段两部分, 两段之间为峡部, 呈90~130°的交角。
近侧段较短, 位于前庭内侧,起始于前庭内侧壁,逐渐向后上方延伸,大部分位于骨迷路的内上方略偏后,与总脚平行;远侧段较长, 相当于“J”的长肢,呈三角形,尖部与峡部相连,末端开口于岩骨后内侧面,呈喇叭形,与内淋巴囊嵌合。
图 5-6:前庭水管的骨迷路及膜迷路相,呈倒“J ”形,近侧段较短, 位于前庭内侧,起始于前庭内侧壁,逐渐向后上方延伸,与总脚平行;远侧段较长, 相当于“J”的长肢,呈三角形,末端开口于岩骨后内侧面,呈喇叭形,与内淋巴囊嵌合。



第40卷第1期中国高原医学与生物学杂志Vol.40No.l 2019年CHINESE HIGH ALTITUDE MEDICINE AND BIOLOGY2019对大前庭水管综合征的基因、影像学诊断研究△马新春,郭斌,徐聪,张英杯(青海大学附属医院耳鼻咽喉科,青海西宁810001)摘要目的探讨SLC26A4基因突变检测及颖骨高分辨率CT检查对诊断大前庭水管扩大综合征的一致性。
方法选取216例人工耳蜗植入的患者作为研究对象,所有患者均行颖骨高分辨率CT检查及基因SLC26A4(2168A>G,IVS7-2A>G)检测。
结果通过两种诊断方法得出大前庭水管扩大综合征的检出率具有一致性(Kappa=0.952)o影像学检查提示在216例患者中,前庭水管扩大者39例;基因检测结果提示基因突变者36例,其中两位点(2168A>G、IVS7-2A>G)的杂合突变16例,纯合突变18例,复合杂合突变2例。
结论SLC26A4基因突变检测与颖骨高分辨率CT检查对诊断大前庭水管扩大综合征具有良好的一致性;基因检测对于可疑前庭水管扩大综合征患者可做到早发现、早诊断、早预防。
关键词:前庭导水管扩大综合征;影像学;责任基因中图分类号:R764.43文献标识码:ADOI:10.13452/ki.jqmc.2019.01.005Diagnosis of large vestibular aqueduct syndrome by geneticdetection and imaging examination A*MA Xin-Chun,GUO Bin,XU Cong,ZHANG Ying(Otolaryngology Of Qing Hai University Affiliated Hospital,QingHai Xining810001) Abstract Objective To explore the consistency of SLC26A4gene mutation detection and temporal bone high resolution CT in diagnosing large vestibular aqueduct syndrome.Methods216patients with cochlear implants were selected as the research objects.All patients underwent the high resolution CT in temporal bone and detect SLC26A4 gene on common sites2168A>G and IVS7-2A>G.Results Two diagnostic methods were used to find that there was consistency in the detection rate of Large vestibular aqueduct syndrome(Kappa二0.952).Imaging examination showed that39cases of vestibular water tube enlarged in216patients.The results of genetic test showed36cases of gene mutation,including16cases of heterozygous mutation at two sites(2168A>G and IVS7-2A>G),18cases of homozygous mutation and2cases of compound heterozygous mutation.Conclusions The two methods have good consistency in the diagnosis of large vestibular aqueduct syndrome.SLC26A4gene mutation detection and high resolution CT examination of the temporal bone have good consistency in the diagnosis of Large vestibular aqueduct syndrome.Gene chip detection can be done for early detection,early diagnosis and early prevention of patients with suspected mutation.Keywords:Large vestibular aqueduct syndrome;Imaging examination;Liability genes△:青海省卫计委基础研究科研项目(2016-wjzdx-64);※:通信作者,硕士生导师,教授,E-mail:guobin.3a@ 马新春( 1965-),男,回族,青海籍,教授,主任医师26高分辨率颍骨轴位CT检查被认为是确诊大前庭水管综合征(large vestibular aqueduct syndrome, LVAS)的金标准''2|,但患儿对该检查配合性较差。
大前庭水管综合征SLC26A4基因突变临床研究山西医科大学硕士学位论文大前庭水管综合征SLC26A4基因突变临床研究姓名:席宏申请学位级别:硕士专业:耳鼻咽喉-头颈外科学指导教师:张芩娜2011-03-15 山西医科大学硕士学位论文大前庭水管综合征 SLC26A4 基因突变分析摘要目的: 应用耳聋基因芯片对非综合征性感音性神经性耳聋患者进行分子病因学研究。
在临床上可以形成一个将临床听力学特征性的表现,与影像学和致病基因的突变检测相结合的一个系统的诊断流程。
研究方法: 采集聋哑学校和门诊散发病例的非综合症性耳聋患者 102 人的外周血,提取基因组DNA,应用耳聋基因芯片检测中国人中常见的大前庭水管综合征基因(亦称 PDS 基因或SLC26A4 基因)的两个热点突变,包括 IVS7-2A〉G 和 2168A〉G。
结果: 在非综合征行耳聋患者中 SLC26A4 基因突变占 16.39,且 SLC26A4 突变的患者 CT 检查均证实为大前庭水管扩大。
且 CT 证实为大前庭的患者,有 95.36基因检测 SLC26A4 突变阳性,SLC26A4 基因中的 IVS7-2A〉G 位点突变的比率高,占 78.95。
结论:本实验102例非综合征性耳聋患者SLC26A4基因突变率达到16.4,证实了SLC26A4是非综合征性耳聋的易感基因。
运用耳聋基因芯片对非综合征性重度和极重度耳聋患者的突变检出率与解放军总医院的相关报告一致(21)。
SLC26A4基因检测可作为重度和极重度非综合征性耳聋患者的临床检查项目。
本课题对非综合征性耳聋患者都采用CT检查与基因芯片筛查,二者吻合率达94.12,说明SLC26A4基因检测可作为重度和极重度非综合征性耳聋患者的临床检查项目。
筛查该基因有助于该病的预防、诊断和治疗。
关键词:大前庭水管综合征 SLC26A4基因突变基因芯片II山西医科大学硕士学位论文 Abstract Objective: we will analyze the auditory data andgenetic characteristics the geneticdiagnostic kit of hereditary deafness based on DNA microarray. Methods: 61 moderate to profound NSHL patients were included in this study. Theirgenomic DNA samples were extracted from peripheral blood and detected with theDNA microarray which is able to perform mutation detection of 2 hot-spot mutationsin the most common pathologic genes SLC26A4 IVS7-2A〉G G 2168A〉simultaneously. Results: Of 61 patients1016.39 were found out to be carriers of SLC26A4 pathogenicgene mutation .Among them IVS7-2A〉G homozygous mutation in9cases compoundheterozygous mutation in 1 cases 2168A〉Gheterozygous mutation in 1cases. Conclusion: In the 61 NSHL patients the SLC26A4 mutation’s quotiety is 16.39. the resultas same as the PLA General Hospital. the genetic diagnostic kit of hereditarydeafness based on DNA microarray appears to have some inherent advantages ingenetic diagnosis of NSHL its features of easy manipulation standardization andpopularization endue it perfect prospect in clinical application. Key words: Large vestibular aqueduct syndrome SLC26A4 gene Mutation DNAmicroarray III 山西医科大学硕士学位论文英文缩写词索引缩写英文全称中文全称NSHL Nonsyndromic Hearing Loss 非综合征性耳聋LVAS Large Vestibular Aqueduct Syndrome大前庭水管综合征EVA Enlargement of the vestibular aqueduct 前庭导水管扩大PDS Pendredsyndrome Pendred 综合征PCR Polymerase Chain Reaction 聚合酶链反应CT Computerized Tomography 电脑断层扫描ABR Auditory Brainstem Response 听觉脑干反应ASSR Auditory Steady StateResponse 听觉稳态反应SLC26A4 Solute carrierfamily 26 member 4 溶质载体家族 26 个,会员 4HHL Hereditary Hearing Loss 遗传性耳聋 IV 学位论文独创性声明本人声明,所呈交的学位论文系在导师指导下本文独立完成的研究成果。
SLC26A4基因与前庭导水管扩大综合征相关性研究
范建辉
【期刊名称】《医学综述》
【年(卷),期】2010(16)13
【摘要】近十年来的国内外研究证实,由SLC26A4基因突变引起的前庭导水管扩大是常见的致聋的先天性疾病.随着基因诊断技术的发展,越来越多的前庭导水管扩大病例被证实与SLC26A4基因突变有关.现就SLC26A4基因与前庭导水管扩大综合征相关性研究现状及其基因筛查的意义予以综述.
【总页数】3页(P1924-1926)
【作者】范建辉
【作者单位】昆明医学院昆明总医院临床学院,昆明,650031;成都军区昆明总医院耳鼻咽喉-头颈外科,昆明,650031
【正文语种】中文
【中图分类】R764
【相关文献】
1.SLC26A4基因突变致前庭水管扩大家系分析 [J], 张帅;张官萍;陆钊群;袁佛良;魏凡钦
2.95例前庭水管扩大核心家系SLC26A4基因特异突变图谱 [J], 赵亚丽;王秋菊;李庆忠;兰兰;袁虎;纵亮;韩明鲲;王大勇;翟所强
3.前庭水管扩大患者人工耳蜗植入及SLC26A4基因的突变分析 [J], 冯永;胡杰;夏昆;贺楚峰;胡浩;梅凌云;贺定华;刘铮铮;陈红胜
4.SLC26A4基因突变致前庭水管扩大听力损失机制的研究进展 [J], 薛文悦; 陈正侬
5.1例前庭水管扩大患儿SLC26A4基因的新变异研究 [J], 胡澜也;陈洁;辛渊;方旭华;焦宇
因版权原因,仅展示原文概要,查看原文内容请购买。
30例大前庭水管综合征患者 SLC26A4基因诊断与听性脑干
电位特性分析
高东梅;刘洪泉;金鹏;于姝媛;王苹;杜波
【期刊名称】《中国实验诊断学》
【年(卷),期】2016(020)009
【总页数】4页(P1586-1589)
【作者】高东梅;刘洪泉;金鹏;于姝媛;王苹;杜波
【作者单位】吉林大学第一医院耳鼻咽喉头颈外科,吉林长春 130021;吉林大学第一医院耳鼻咽喉头颈外科,吉林长春 130021;吉林大学第一医院耳鼻咽喉头颈外科,吉林长春 130021;吉林大学第一医院耳鼻咽喉头颈外科,吉林长春130021;吉林大学第一医院耳鼻咽喉头颈外科,吉林长春 130021;吉林大学第一医院耳鼻咽喉头颈外科,吉林长春 130021
【正文语种】中文
【相关文献】
1.大前庭水管综合征患者的SLC26A4基因诊断 [J], 梁爽;王树峰
2.河南地区青年和儿童感音神经性聋患者中大前庭水管综合征的临床和基因诊断[J], 能玲玲;董明敏
3.SLC26A4基因与前庭导水管扩大综合征相关性研究 [J], 范建辉
4.双侧大前庭水管综合征患儿的前庭诱发肌源性电位特征 [J], 张玉忠;许珉;任晓勇;张青;张滟;魏馨雨;陈耔辰;徐勇;成颖;高滢;陈飞云;胡娟
5.大前庭水管综合征的基因诊断和SLC26A4基因突变分析 [J], 戴朴;韩东一;冯勃;康东洋;刘新;袁慧军;曹菊阳;张昕;翟所强;杨伟炎;吴柏林
因版权原因,仅展示原文概要,查看原文内容请购买。
大前庭水管综合征及其相关的基因—SLC26A4赵亚丽翟所强王秋菊中国人民解放军总医院耳鼻咽喉研究所(北京100853)临床和流行病学研究表明,每1000名新生儿中就有1名先天性耳聋的患儿。
大前庭水管综合征(largevestibularaqueductsyndrome,LVAS)是先天性耳聋的一种。
1978年,Valvassori和Clemis[1]在对3700例颞骨连续分层摄片中发现,有50例前庭水管扩大,并将其命名为大前庭水管(dilatedvestibularaqueduct,DVA),又将临床上伴感音神经性听力损失等症状者称“大前庭水管综合征”。
近年,随着CT、MRI以及分子遗传学的发展,对此病的认识已经从影像学诊断、临床表现、发展到了分子诊断的水平。
1大前庭水管综合征的临床表现、发病机理及诊断大前庭水管是儿童感音神经性聋最常见的内耳畸形,由其导致的大前庭水管综合征是一种独立性疾病,除了前庭水管扩大外,不合并其它畸形,属于非综合征型耳聋。
其临床表现为波动性感音神经性听力损失,多呈进行性下降。
可从出生后至青春期这一年龄段内任何时期开始起病,但多数为出生后几年内发病。
发病突然或隐匿,通常之前有感冒、轻微颅外伤或其他使颅内压增高的病史。
约81%-94%的患者为双侧发病,单侧发病者少见[2]。
听力受损以高频为主,听力图多为下降型,少数为平坦型,无一例为上升型[3]。
听力下降的程度与前庭水管的大小无关,可表现为从接近正常到极重度聋。
并且,双耳听力可不对称。
大多数患者仅表现为听力损失,只有少数患者伴有前庭症状,约占29%,但有人认为前庭症状少见可能与发病人群多为幼儿有关[4]。
目前,关于前庭水管扩大导致感音神经性聋的致病机理还不是很清楚,通常有三种假说:(1)内淋巴液返流学说,前庭水管扩大时,往往同时伴有内淋巴管和内淋巴囊的异常扩大,突然的脑压变化,迫使两层脑膜间内淋巴囊内容返流入耳蜗,囊内高渗内淋巴进入耳蜗基底周,损伤神经感觉上皮,产生感音神经性耳聋。
(2)膜迷路破裂和外淋巴漏学说,LVAS膜迷路较菲薄,在基底膜和前庭膜处可产生蜗内破裂使内外淋巴液混合,损伤毛细胞,产生进行性感音神经性聋。
(3)脑压波动殃及内耳,脑脊液压力异常波动,经宽大的前庭水管传到内耳,正常情况脑压变化,被狭窄的前庭水管和耳蜗水管缓冲,当前庭水管扩大耳蜗水管正常时,快速脑压变化使耳蜗暂时压力失衡,造成膜迷路损伤或蜗内漏管。
大多数学者赞同第一种假说。
并且,内淋巴囊内淋巴液比耳蜗内淋巴液蛋白成分高十几倍,倒流于蜗管,可损害毛细胞而出现耳聋[5]。
LVAS的诊断依据主要是:结合儿童有感冒、头部外伤或其他使颅内压增高的病史,听力检查示高频听力下降为主的感音神经性聋,高分辨率轴位CT扫描可见,颞骨岩部后缘有一深且大的三角形骨质缺损影,缺损边缘清晰,内端连接前庭或总脚,前庭水管中点(半规管总脚至前庭水管外口间1/2处)的宽度大于1.5mm。
近年,Okamoto等[6]提出,前庭水管扩大的病例几乎均有内淋巴管和内淋巴囊的扩大。
因此又给LVAS的诊断增加了一条途径。
2大前庭水管综合征相关基因的研究近10年来,随着分子遗传学技术的发展和人类连锁图谱的绘制,使发现遗传性耳聋相关基因、破译听觉系统的遗传基因编码、探索遗传性耳聋发病机理的研究产生了革命性的飞跃。
对于LVAS的分子学病因的研究也在不断开展。
1996年,Griffith首先[7]报道了LVAS男性先证者和其患同一疾病之・聋病基因专辑・基金项目:国家自然基金面上项目(30370782&30470956),北京市重大科技专项子课题(H020220020610)及“863”计划滚动项目(2004AA221080)联合资助。
作者简介:赵亚丽(1980-),女,河北人,医学硕士,专业方向:遗传性耳聋基因的分子流行病学研究通讯作者:王秋菊,副教授。
E-mail:wqcr@301ent.org兄弟出生于无耳疾的父母,嗣后Nowake等[8]报道姐弟同患LVAS的病例。
1997年,Abe等[9]推测,LVAS是以常染色体隐性遗传的方式遗传的。
1999年,Usami等[10]把前庭水管扩大伴感音神经性聋的非综合征性聋的基因定位在7q31,与Pendred综合征的致病基因相同,即SLC26A4,又名PDS基因。
Usami等认为SLC26A4基因突变,可引起综合征或非综合征性感音神经性聋,LVAS为非综合征性的。
下面对SLC26A4基因的结构、功能、突变谱与致病机理作一阐述。
2.1SLC26A4基因的结构Everett等[11]应用GRAIL生物信息分析软件,将SLC26A4cDNA序列与同源BACRG364P16序列进行比较,推断SLC26A4基因的mRNA全长4930bp,含外显子21个,开放阅读框架2343bp,每个内含子与外显子的界限分明,众多的外显子表明SLC26A4基因将编码一个结构和功能十分复杂的蛋白质。
SLC26A4基因的开放阅读框架起始于2号外显子,穿越剩余20个外显子。
除21号外显子之外,其它外显子长度约为55-231bp。
Haila等[12]发现,SLC26A4基因与DRA(downregulatedinadenomd)基因具有高度同源性,两者外显子数目相同,对应外显子序列长度相近。
但对应内含子的序列长度却差异较大。
这种相似的外显子序列反映两种基因编码的蛋白质功能相似,内含子之间的差异意义尚待进一步阐明。
2.2SLC26A4基因的表达分布与功能用NorthernBlot分析显示,SLC26A4基因在甲状腺高表达,在成年人和胎儿的肾脏及脑组织,仅有弱的表达。
SLC26A4基因在甲状腺的高表达说明其与甲状腺功能有关。
有人用PCR分析胎儿耳蜗cDNA文库,检测到SLC26A4基因在耳蜗内的表达。
1999年,Everett等[13]利用RNA的原位杂交技术,观察了从胚胎8天到出生后5天小鼠内耳SLC26A4基因mRNA的表达模式。
发现在胚胎13天,小鼠内淋巴管和内淋巴囊就有mRNA的强表达,而在胚胎15天时,椭圆囊、球囊和耳蜗才有弱表达,这就说明SLC26A4基因的突变可能导致SLC26A4基因的高表达区—内淋巴管和内淋巴囊的功能障碍。
SLC26A4基因编码的是一个分子量为86kD,含780个氨基酸的蛋白质,即Pendrin。
Pendrin主要是由疏水性氨基酸组成,其与DRA(downregulatedinadenomd)基因编码的蛋白具有高度的同源性,同属于离子转运体家族。
因此,Everett等[11]推测Pendrin可能是一种硫酸盐转运子。
1999年,Scott等[14]利用显微注射的方法把SLC26A4基因的cRNA注入爪蟾卵母细胞内,并使其表达Pendrin,结果发现,与对照组相比较,其硫酸盐的转运功能没有明显差异,而碘和氯的转运较对照组分别增加30倍和10倍。
此外,Scott等还对SLC26A4基因重组的杆状病毒转染的Sf9细胞进行研究,也得到类似的结果,提示Pendrin并不是一种硫酸盐转运子,而是一种碘/氯离子转运子。
为了进一步研究Pendrin在细胞的定位,Bidart等[15]用抗肽抗体和实时定量PCR技术,研究了人体甲状腺组织在正常、良性和恶性病变时Pendrin的表达,结果发现,仅在甲状腺滤泡细胞的顶膜存在Penrin表达。
Royaux等用特异性多肽抗体对Pendrin进行免疫定位,也发现Pendrin主要位于甲状腺滤泡上皮细胞顶端亚膜单位。
他认为,当碘离子从基底膜侧吸收到甲状腺滤泡细胞后,Pendrin及时把碘离子转运到滤泡胶质内,与甲状腺过氧化物酶作用导致碘的有机化,并与甲状腺球蛋白结合,维持甲状腺的正常功能。
既然Pendrin不仅在甲状腺表达,也可在内耳的内淋巴管、内淋巴囊及Corti’s器外沟细胞中表达(这些结构与内淋巴液代谢有关),因此Pendrin可能作为氯离子转运体,调节内淋巴液的离子平衡[13]。
2.3SLC26A4基因突变与表型变化SLC26A4基因的突变可以导致DVA、Mondini畸形和甲状腺肿,如此多变的表型预示SLC26A4存在一个广泛的基因突变谱。
目前已经报道的SLC26A4基因的突变位点超过100个,其中绝大部分是错义突变,另外还有框移突变和剪接位点突变。
这些突变在mRNA链上的分布无明显聚集现象,除外显子20尚未发现突变外,其余各个外显子上均检测到突变位点。
在对西方人群的研究中,最常见的突变位点是L236P,其曾经在调查的26个家庭[16-21]中筛查到,它位于外显子6上,编码的蛋白位于细胞外第三个环上。
其次分别是T416P、H723R、IVS8+1G>A,分别位于外显子10、外显子19和内含子8上,除T416P位于氨基酸序列的高度保守区外,H723R和L236P均位于非保守区[16],IVS8+1G>A位于外显子8的剪接位点,其突变影响前体mRNA的剪接过程,从而影响Pendrin的结构。
在众多的突变中,多数突变既见于Pendred综合征,又见于LVAS。
因此,同一位点的突变可能导致不同的临床表现。
SLC26A4基因在甲状腺和胚胎组织的内耳均有表达,但Pendrin蛋白是如何使这样两个完全不同的组织同时致病的呢?如前所述,Pendrin在甲状腺是表达在滤泡细胞的顶膜,当Pendrin蛋白功能障碍时,不能把碘离子及时转运到滤泡胶质,使碘离子在滤泡细胞内积聚,从而不能有效地有机化与甲状腺球蛋白结合,这一点与Pendred综合征的病人的甲状腺球蛋白水平多数升高相一致,可能是负反馈作用的结果。
但多数Pendred综合征病人血中的甲状腺激素水平是正常的,只有少数人轻度升高,机制尚不清楚。
1999年,Qvortrup等[22]曾报道,大鼠的内淋巴囊类似甲状腺滤泡,有平衡胶状物质填塞囊腔,内淋巴囊上皮细胞的功能类似于甲状腺滤泡细胞,可以合成、分泌、吸收及消化蛋白质。
因此,Pendrin可能在维持内淋巴平衡上发挥作用,此学说有可能解释同一种蛋白功能缺陷以导致两种不同组织疾病。
在内耳,氯离子转运障碍可导致内淋巴液成分的改变,从而损伤感觉上皮细胞,且由于渗透压改变和成分改变的毒性机制导致膜迷路结构改变。
由于内淋巴管(ED)和内淋巴囊(ES)到4岁时才停止发育,因此它们的扩大可导致周围骨性结构的改变,如前庭水管或耳蜗结构的改变[23]。
因此,内淋巴管和内淋巴囊的扩大对于LVAS和Penred综合征都很有诊断价值。
为了深入研究突变的SLC26A4基因编码的Pendrin的致病机理,Taylor等[24]观察了9个(L117F、Q446R、V138F、T410M、Y556C、G672E、G209、G102R、L236P)SLC26A4基因的错义突变的突变体在细胞的定位和功能上的变异。