原位杂交探针大体可分为三类:寡核苷酸探针,cDNA探针,RNA探针。寡核苷酸探针由25bp左右的核苷酸组成,通常可以由公司合成,由于其序列较短,因此特异性较强,原位杂交时可以区分家族基因,同一基因的不同剪切体,cDNA探针长约500bp左右,可以通过非对称PCR扩增合成,由于探针式DNA,可以有效的避免降解,但DNA和RNA的结合不如RNA与RNA结合强,通常不采用,RNA探针长约500bp左右,通过体外转录合成,如果设计合理,其特异性是可以得到保证的,其主要缺点是容易被RNAse酶降解。
查看原位杂交相关的文献发现,2000年以前的原位杂交常使用放射性标记的寡核苷酸探针,通过胶片曝光来显色,2000年以后的文献常使用RNA探针。许多肾脏发育的文献虽然有很多原位杂交数据,但却没有附带上探针序列或者用于探针模板克隆的引物,通过了解厦门黄老师斑马鱼和昆明毛炳宇爪蟾中原位杂交探针设计方法,我们可以采用RNA探针。文献中很难找到关于RNA探针设计的原则,有的文献报道直接用cDNA合成RNA探针。借鉴爪蟾中原位杂交探针设计流程,以小鼠FGF10的探针设计为例进行介绍。
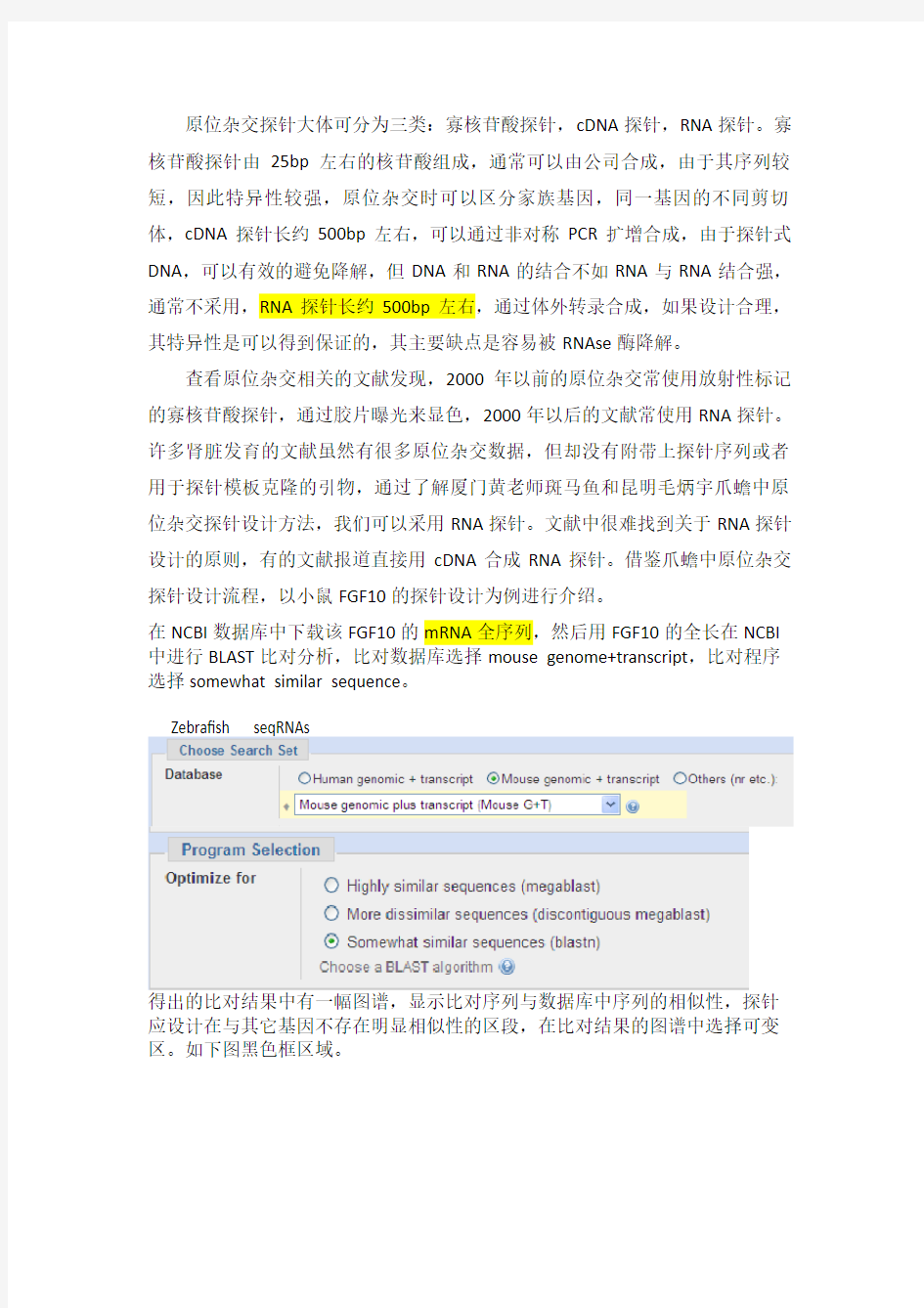
在NCBI数据库中下载该FGF10的mRNA全序列,然后用FGF10的全长在NCBI 中进行BLAST比对分析,比对数据库选择mouse genome+transcript,比对程序选择somewhat similar sequence。
Zebrafish seqRNAs
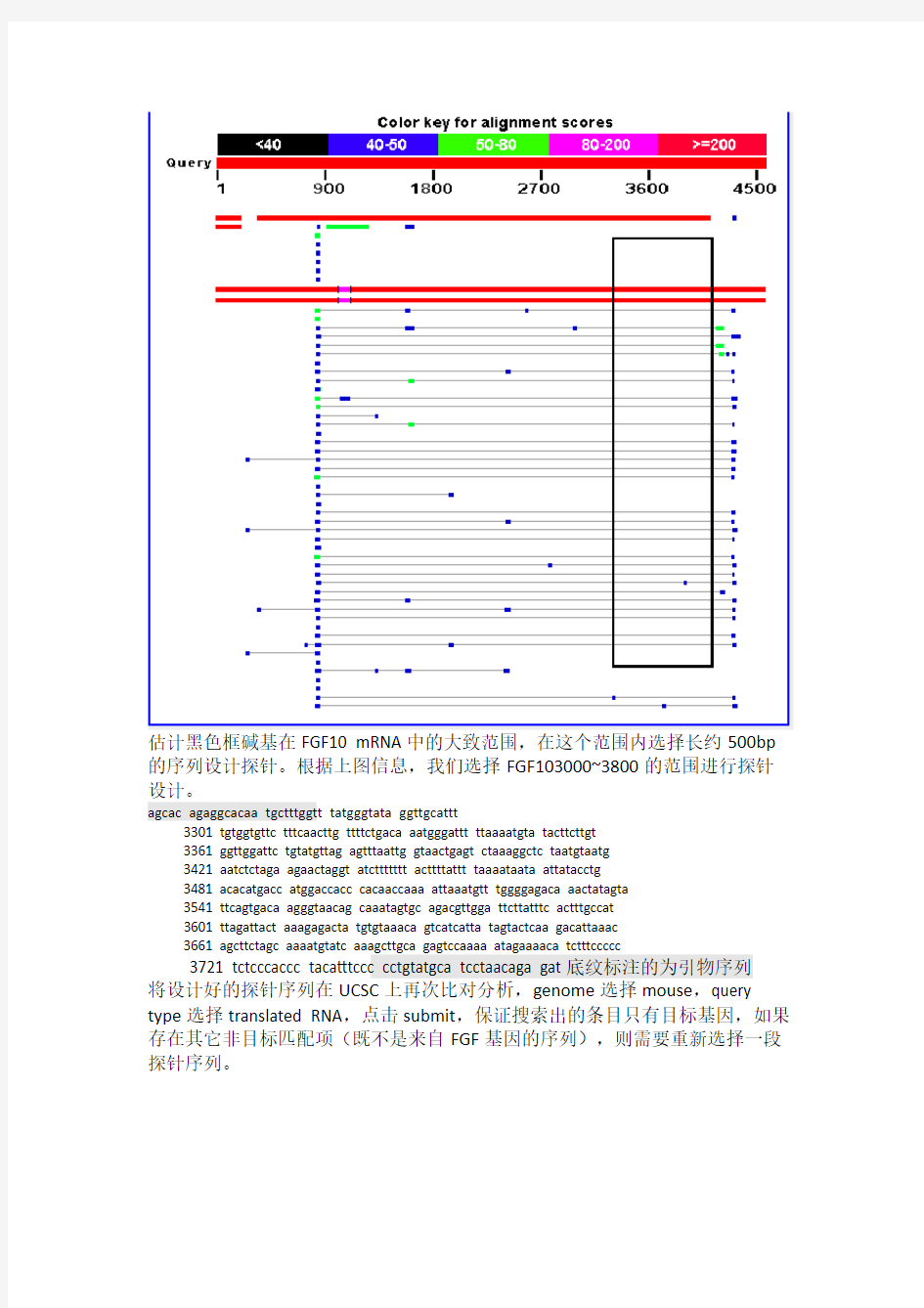
得出的比对结果中有一幅图谱,显示比对序列与数据库中序列的相似性,探针应设计在与其它基因不存在明显相似性的区段,在比对结果的图谱中选择可变区。如下图黑色框区域。
估计黑色框碱基在FGF10 mRNA中的大致范围,在这个范围内选择长约500bp 的序列设计探针。根据上图信息,我们选择FGF103000~3800的范围进行探针设计。
agcac agaggcacaa tgctttggtt tatgggtata ggttgcattt
3301 tgtggtgttc tttcaacttg ttttctgaca aatgggattt ttaaaatgta tacttcttgt
3361 ggttggattc tgtatgttag agtttaattg gtaactgagt ctaaaggctc taatgtaatg
3421 aatctctaga agaactaggt atcttttttt acttttattt taaaataata attatacctg
3481 acacatgacc atggaccacc cacaaccaaa attaaatgtt tggggagaca aactatagta
3541 ttcagtgaca agggtaacag caaatagtgc agacgttgga ttcttatttc actttgccat
3601 ttagattact aaagagacta tgtgtaaaca gtcatcatta tagtactcaa gacattaaac
3661 agcttctagc aaaatgtatc aaagcttgca gagtccaaaa atagaaaaca tctttccccc
3721 tctcccaccc tacatttccc cctgtatgca tcctaacaga gat底纹标注的为引物序列
将设计好的探针序列在UCSC上再次比对分析,genome选择mouse,query type选择translated RNA,点击submit,保证搜索出的条目只有目标基因,如果存在其它非目标匹配项(既不是来自FGF基因的序列),则需要重新选择一段探针序列。
你可以点击browser浏览一下所设计的探针序列是否是FGF10的序列以及所在位置。
在选择探针序列时,尽量选择3’端的区域,避开可能的选择性剪切,同时也要避免mRNA的末端序列,因为可能存在翻译终止序列,影响以后的体外转录效率。经过上述分析后,探针序列可以较好的避免家族基因的保守序列,确保特异性。
我们可以针对每个基因设计两个探针,后面比较一下哪个探针杂交效果最好。
原位杂交试剂 1.Buffer I溶液 试剂剂量 Tris-HCl 7.882g NaCl 4.4g 纯水至500ml 混匀,调pH至7.5,室温保存。 2.Buffer Ⅱ溶液 试剂剂量 Buffer I溶液10ml Blocking(索莱宝公司) 0.1g
调pH至8.0,水浴溶解,过滤除菌分装,-20℃保存。 3.Buffer Ⅲ溶液 试剂剂量 Tris-HCl 7.882g NaCl 3g MgCl2·6H2O 5.1g 纯水至500ml 混匀,调pH至9.5,室温保存。 4.2×SSC 试剂剂量 20×SSC 100ml 纯水至1000ml 混匀,室温保存。 5.1×SSC 试剂剂量 20×SSC 50ml 纯水至1000ml 混匀,室温保存。 6.0.1×SSC 试剂剂量 1×SSC 100ml 纯水至1000ml 混匀,室温保存。 7.蛋白酶K稀释液 试剂剂量 Tris-HCl 7.882g EDTA 7.3g 纯水至500ml 混匀,调pH至8.0,室温保存。 原位杂交 1.探针构建 由上海生工生物工程有限公司合成本实验所用的探针。合成好的探针粉末用无核酶水溶解,浓度为0.5μg/μl。
2.探针标记 探针标记体系如下(10μl): 试剂剂量 合成的探针2μl Dig RNA 1μl 10×Buffer缓冲液1μl RNA聚合酶(SP6/T7)1μl DTT(100mM)1μl RI 0.5μl 无核酶水至10μl 混匀,37℃水浴1h。 后加入2μl DNA酶I,37℃水浴15min,降解模板DNA。再加入2μl EDTA (0.2M)终止反应。加入1/10体积的LiCl(4M)及2.5倍体积预冷的无水乙醇,混匀后置于-20℃冰箱过夜、沉淀。第二天,4℃,12000×g离心15min,弃去上清。用预冷的75%乙醇洗一次,4℃,12000×g离心1min,吸尽上清,晾干。溶于50μl无核酶水中,待完全溶解后分装,-80℃保存。 3.斑点杂交 1)从-80℃冰箱中取出已经分装好的探针原液,置于80℃水浴锅中热激5min,使探针变性,然后立即插入冰中。 2)用探针稀释液将变性后的探针依次稀释10、20、50倍。 3)取探针原液及稀释液各1μl依次点在做好标记的尼龙膜上,下面垫一张空膜。 4)将尼龙膜置于紫外仪中交联30s,再放入Buffer I缓冲液中平衡5min。 5)用BufferⅡ在室温条件下封闭1h,使用BufferⅡ稀释地高辛抗体(1:1000)室温孵育2h或者4℃过夜。 6)用BufferI洗膜3次,每次15min,后置于BufferⅢ中平衡5min,再倒掉。 7)用BufferⅢ稀释NBT/BCIP作为显色剂,稀释比例为1:50。 8)避光,滴加稀释好的显色剂,孵育10-20min,显色,根据显色结果选择合适的探针浓度。 4.原位杂交 原位杂交过程中需要全程在无核酶的环境中进行,所有用到的实验物品都需要经过无核酶处理方可使用。 1)从培养箱中取出细胞,室温静置平衡5min,用PBS洗2次,每次3min,晾干。 2)4%PFA固定20min,PBS洗3次,每次5min,晾干。
第一章材料 SPCC 一般用钢板,表面需电镀或涂装处理 SECC 镀锌钢板,表面已做烙酸盐处理及防指纹处理 SUS 301 弹性不锈钢 SUS304 不锈钢 镀锌钢板表面的化学组成------基材(钢铁),镀锌层或镀镍锌合金层,烙酸盐层和有机化学薄膜层. 有机化学薄膜层能表面抗指纹和白锈,抗腐蚀及有较佳的烤漆性. SECC的镀锌方法 热浸镀锌法: 连续镀锌法(成卷的钢板连续浸在溶解有锌的镀槽中 板片镀锌法(剪切好的钢板浸在镀槽中,镀好后会有锌花. 电镀法: 电化学电镀,镀槽中有硫酸锌溶液,以锌为阳极,原材质钢板为阴极. 1-2产品种类介绍 1.品名介绍 材料规格后处理镀层厚度 S A B C*D*E S for Steel A: EG (Electro Galvanized Steel)电气镀锌钢板---电镀锌 一般通称JIS 镀纯锌EG SECC (1) 铅和镍合金合金EG SECC (2) GI (Galvanized Steel) 溶融镀锌钢板------热浸镀锌 非合金化GI,LG SGCC (3) 铅和镍合金GA,ALLOY SGCC (4) 裸露处耐蚀性2>3>4>1 熔接性2>4>1>3 涂漆性4>2>1>3 加工性1>2>3>4
B: 所使用的底材 C (Cold rolled) : 冷轧 H (Hot rolled): 热轧 C: 底材的种类 C: 一般用 D: 抽模用 E: 深抽用 H: 一般硬质用 D: 后处理 M: 无处理 C: 普通烙酸处理---耐蚀性良好,颜色白色化 D: 厚烙酸处理---耐蚀性更好,颜色黄色化 P: 磷酸处理---涂装性良好 U: 有机耐指纹树脂处理(普通烙酸处理)--- ---耐蚀性良好,颜色白色化,耐指纹性很好A: 有机耐指纹树脂处理(厚烙酸处理)---颜色黄色化,耐蚀性更好 FX: 无机耐指纹树脂处理---导电性 FS: 润滑性树脂处理---免用冲床油 E: 镀层厚 1-4物理特性 膜厚---含镀锌层,烙酸盐层及有机化学薄膜层,最小之膜厚需0.00356mm以上. 测试方法有磁性测试(ASTM B499), 电量分析(ASTM B504), 显微镜观察(ASTM B487) 表面抗电阻---一般应该小于0.1欧姆/平方公分. 1- 5 盐雾试验----试片尺寸100mmX150mmX1.2mm, 试片需冲整捆或整叠铁材中取下,必须在镀烙酸盐后24小时,但不可超过72小时才可以用于测试,使用5%的盐水,用含盐的水汽充满箱子,试片垂直倒挂在箱子中48小时。 测试后试片的镀锌层不可全部流失,也不能看到底材或底材生锈,但是离切断层面6mm范围有生锈情况可以忽略。
引物探针设计简介 已有2993 次阅读2009-1-1 20:48|个人分类:课堂集锦|系统分类:科研笔记 1.寡聚核苷酸引物的选择,通常是整个扩增反应成功的关键。所选的引物序列将决定PCR 产物的大小、位置、以及扩增区域的Tm值这个和扩增物产量有关的重要物理参数。好的引物设计可以避免背景和非特异产物的产生,甚至在RNA-PCR中也能识别cDNA或基因组模板。引物设计也极大的影响扩增产量:若使用设计粗糙的引物,产物将很少甚至没有;而使用正确设计的引物得到的产物量可接近于反应指数期的产量理论值。当然,即使有了好的引物,依然需要进行反应条件的优化,比如调整Mg2+浓度,使用特殊的共溶剂如二甲基亚砜、甲酰胺和甘油。计算机辅助引物设计比人工设计或随机选取更有效。一些影响PCR反应中引物作用的因素诸如溶解温度、引物间可能的同源性等,易于在计算机软件中被编码和限定。计算机的高速度可完成对引物位置、长度以及适应用户特殊条件的其他有关引物的变换可能性的大量计算。通过对成千种组合的检测,调整各项参数,可提出适合用户特殊实验的引物。因此通过计算机软件选择的引物的总体“质量”(由用户在程序参数中设定)保证优于通过人工导出的引物。需要指出的是,引物不必与模板完全同源,因此可包含启动子序列、限制酶识别位点或5'端的各种修饰,这种对引物的修饰不会妨碍PCR反应,而会在以后使用扩增子时发挥作用。 2.基本PCR引物设计参数引物设计的目的是在两个目标间取得平衡:扩增特异性和扩增效率。特异性是指发生错误引发的频率。特异性不好或劣等的引物会产生额外无关和不想要的PCR扩增子,在EB染色的琼脂糖凝胶上可见到;引物效率是指在每一PCR循环中一对引物扩增的产物与理论上成倍增长量的接近程度。①引物长度;特异性一般通过引物长度和退火温度控制。如果PCR的退火温度设置在近于引物Tm值(引物/模板双链体的解链温度)几度的范围内,18到24个碱基的寡核苷酸链是有很好的序列特异性的。引物越长,扩增退火时被引发的模板越少。为优化PCR反应,使用确保溶解温度不低于54℃的最短的引物,可获得最好的效率和特异性。总的来说,最好在特异性允许的范围内寻求安全性。每增加一个核苷酸,引物特异性提高4倍;这样,大多数应用的最短引物长度为18个核苷酸。引物设计时使合成的寡核苷酸链(18~24聚物)适用于多种实验条件仍不失为明智之举。②引物的二级结构包括引物自身二聚体、发卡结构、引物间二聚体等。这些因素会影响引物和模板的结合从而影响引物效率。对于引物的3'末端形成的二聚体,应控制其ΔG大于
原位杂交 1 探针的设计与合成 1)根据实验室已有的p8基因cDNA全长序列,用premier primer5.0设计引物p81和p82, 以卤虫cDNA为模板,PCR扩增得到346bp的产物,用Takara胶回收试剂盒回收纯化。引物编号引物序列长度 p81 TGCGGACGAAACAGGAAG 18 bp p82 GCTCAAACAGTGA TGCCAGT 20 bp 2)目的片段克隆 a. 在无菌离心管中加入连接载体的各种成分,载体与片段的摩尔比控制在1:3-1:8,根据凝胶电泳检测后的浓度及载体与片段分子大小来计算摩尔比。加入成分及比例如下: 目的PCR片段 5 μl pGM-T载体(约50ng/uL) 1 μl 10×T4 DNA Ligation Buffer 1 μl T4 DNA Ligase(3U/uL) 1 μl 无菌去离子水 3 μl 总体积10 μl b. 轻轻弹动离心管以混合内容物,短暂离心。置于PCR仪中16℃过夜连接,反应结束后将离心管置于冰上。 c. 向铺好的含有氨苄青霉素的固体平板表面加入16 μl的IPTG(50mg/ml)、40 μl的X-gal (20mg/ml),使用无菌的弯头玻璃棒将其均匀的涂开,避光置于37℃培养箱1-3小时,使溶解X-gal的二甲基甲酰挥发干净。 d. 将10 μl的连接产物加到100 μl DH5 感受态细胞中,轻弹混匀,冰浴半小时,将离心管置于42℃水浴90秒,取出管后立即置于冰浴上放置2-3分钟,其间不要摇动离心管。向离心管加入500 μl 37℃预热的LB(不含抗生素)培养基,150rpm摇床37℃振荡培养45分钟。目的是使质粒上相关的抗性标记基因表达,使菌体复苏。将菌液于4000g下离心10分钟,去掉上清,加入100 μl培养液重溶并加入到配制好的LB固体培养基上,用无菌的弯头玻璃棒轻轻将细胞均匀涂开。待平板表面干燥后,倒置平板,37℃培养12-16小时。 e. 挑取白色菌落直接进行PCR检测,筛选转化子。 f. 将转化子接种于LB液体培养基中培养24小时,吸取1mL菌液送至大连宝生物公司进行序列测序。 3)重组质粒的线性化 取6 μl以测序的重组质粒,选取NcoI内切酶37℃酶切4h。酶切反应体系为20 μl: 质粒 6 μl 10xK Buffer 2 μl NcoI酶 1 μl 0.1% BSA 2 μl 灭菌水9 μl 终体积20 μl 取酶切前后的质粒各4 μl,经1%琼脂糖电泳检测,确认酶切完全,将酶切产物用Takara胶回收试剂盒回收纯化,作为探针合成的模板。 4)探针合成 按罗氏DIG RNA Labeling Kit (SP6/T7)试剂盒使用指南,标记反义RNA探针。 使用的所有试剂和器皿均经去RNase处理,合成方法如下:先准备反应体系。冰上向RNase-Free的微离心管中顺序加入下列试剂:
原位杂交原理及具体操作
原位杂交实验原理与方法 一、目的 本实验的目的是学会原位杂交的使用方法。了解各种原位杂交的基本原理和优缺点。 二、原理 原位杂交组化(简称原位杂交,in situ hybridization histochemistry;ISHH)属于分子杂交的一种,是一种应用标记探针与组织细胞中的待测核酸杂交,再应用标记物相关的检测系统,在核酸原有的位置将其显示出来的一种检测技术。原位杂交的本质就是在一定的温度和离子浓度下,使具有特异序列的单链探针通过碱基互补规则与组织细胞内待测的核酸复性结合而使得组织细胞中的特异性核酸得到定位,并通过探针上所标记的检测系统将其在核酸的原有位置上显示出来。 当然杂交分子的形成并不要求两条单链的碱基顺序完全互补,所以不同来源的核酸单链只要彼此之间有一定程度的互补顺序(即某种程度的同源性)就可以形成杂交双链。 探针的种类按所带标记物可分为同位素标记探针和非同位素标记探针两大类。目前,大多数放
射性标记法是通过酶促反应将标记的基因掺入DNA中,常用的同位素标记物有3H、35S、125I 和32P。同位素标记物虽然有灵敏性高,背底较为清晰等优点,但是由于放射性同位素对人和环境均会造成伤害,近来有被非同位素取代的趋势。非同位素标记物中目前最常用的有生物素、地高辛和荧光素三种。 探针的种类按核酸性质不同又可分为DNA探针、cDNA探针、cRNA探针和合成寡核苷酸探针。cDNA 探针又可分为双链cDNA探针和单链cDNA探针。原位杂交又可分为菌落原位杂交和组织原位杂交。 菌落原位杂交(Colony in situ hybridization)菌落原位杂交是将细菌从培养平板转移到硝酸 纤维素滤膜上,然后将滤膜上的菌落裂菌以释出DNA。将NDA烘干固定于膜上与32P标记的探针杂交,放射自显影检测菌落杂交信号,并与平板上的菌落对位。 组织原位杂交(Tissue in situ hybridization)组织原位杂交简称原位杂交,指组织或细胞的原位杂交,它与菌落的原位杂交不同。菌落原位杂交需裂解细菌释出DNA,然后进行杂交。而原位
real time PCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价 一、实时荧光Taqman 探针设计 总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。即real-time PCR的扩增片段是50bp----150bp。当找不到150bp的保守片段时,必须确保探针的片段是保守的。 在设计探针和引物时,要同时考虑在两条链上设计引物与探针。但要注意的是:在那条链上设计探针时,就应靠近在同一条链上设计的引物(即上游引物)。这样,可保证在将来扩增时,即便没有完全扩增,也有荧光信号报告出来。两者的距离最好是探针的5’端离上游引物的3’有一个碱基,但也可以重叠。 若在原序列中找不到合适的探针与引物(1主要是探针和上游引物的距离太远,而离下游引物的距离却较近时;2突变位点要求在探针的5’ 端也能检测到荧光信号,但却是在3’端),可在互补的序列中设计引物与探针。 另real-time PCR中的探针和引物的Tm值,均要高于平常PCR的引物和杂交的探针的Tm值。 二、探针的设计 探针设计的基本原则: 1.保守:探针要绝对的保守,有时分型就单独依靠探针来决定。理论上有一个碱基不配对,就可能检测不出来。若找不到完全保守的片段,也只能选取有一个碱基不同的片段。且这个不同的碱基最好在探针的中间,对探针与目的片段的杂交影响不大,不相同的碱基最好不要在两端,因为两端不利于探针的杂交。且最好为A或T,而不能为G或A,因为A、T为双键,而G、A为三键。 2.探针长度
Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,最好为70℃,确保探针的Tm 值要比引物的Tm值高出10℃,这样可保证探针在煺火时先于引物与目的片段结合。因此探针最好是富含GC的保守片段,保证其的Tm值较高。现在有Taqman MGB探针,在TAMER之后再标记一个MGB,可使探针的Tm值较高,即使探针片段较短,也可达到Taqman探针的Tm值要求(68-70℃)。 3.探针的名称 应标记探针在基因组的位置及长度。 4.探针Tm值计算 用oligo或primer preiemer软件即可计算Tm值。确保探针中GC含量在30-80%。应避免探针中多个重复的碱基出现,尤其是要避免4个或超过4个的G碱基出现。 5.探针的评价 用DNAstar软件中的Primerselect软件,点击“log”菜单中的“create primer catalog”,在“name” 中输入探针的名称、位置,按Tab键进入“sequence”,粘贴或输入要分析的探针序列。选中整个序列后,在“report”菜单下“primer self dimer”,分析探针的二聚体。弹出的窗口中就告诉此探针有多少个dime r,并对此探针用dG值进行评价(通常给出最差的dG值,理论上是dG值越大越好)。在“report”菜单下“p rimer hairpins”,分析探针的发夹结构。弹出的窗口中就告诉此探针有多少个hairpins,并对此探针的h airpins进行评价。多重荧光PCR时,要对多条探针进行“pair dimer”进行分析。 6.探针的5’端不能为G 因为即使单个G碱基与FAM荧光报告基团相连时,G可以淬灭FAM基团所发出的荧光信号,从而导致假阴性的出现。 7.Taqman探针与引物之间的位置
1.引物最好在模板cDNA的保守区内设计。 DNA序列的保守区是通过物种间相似序列的比较确定的。在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。 2.引物长度一般在15~30碱基之间。 引物长度(primer length)常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。 3.引物GC含量在40%~60%之间,Tm值最好接近72℃。 GC含量(composition)过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。有效启动温度,一般高于Tm值5~10℃。若按公式Tm= 4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm 值最好接近72℃以使复性条件最佳。 4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。 5.引物3′端不能选择A,最好选择T。 引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C 错配的引发效率介于A、T之间,所以3′端最好选择T。 6. 碱基要随机分布。 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。 7. 引物自身及引物之间不应存在互补序列。 引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。这种二级结构会因空间位阻而影响引物与模板的复性结合。引物自身不能有连续4个碱基的互补。 两引物之间也不应具有互补性,尤其应避免3′ 端的互补重叠以防止引物二聚体(Dimer与Cross dimer)的形成。引物之间不能有连续4个碱基的互补。 引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal/mol)。否则易导致产生引物二聚体带,并且降低引物有效浓度而使PCR 反应不能正常进行。 8. 引物5′ 端和中间△G值应该相对较高,而3′ 端△G值较低。 △G值是指DNA 双链形成所需的自由能,它反映了双链结构内部碱基对的相对稳定性,△G 值越大,则双链越稳定。应当选用5′ 端和中间△G值相对较高,而3′ 端△G值较低(绝对值不超过9)的引物。引物3′ 端的△G 值过高,容易在错配位点形成双链结构并引发DNA 聚合反应。(不同位置的△G值可以用Oligo 6软件进行分析) 9.引物的5′端可以修饰,而3′端不可修饰。 引物的5′ 端决定着PCR产物的长度,它对扩增特异性影响不大。因此,可以被修饰而不影响扩增的特异性。引物5′ 端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入点突变、插入突变、缺失突变序列;引入启动子序列等。引物的延伸是从3′ 端开始的,不能进行任何修饰。3′ 端也不能有形成任何二级结构可能。 10. 扩增产物的单链不能形成二级结构。
荧光原位杂交(FISH)探针的制备及其应用 概述 1、克隆性染色体异常是肿瘤的特征 2、染色体异常常见的类型 3、染色体异常的检测方法 二、荧光原位杂交及其探针 1、荧光原位杂交的原理 2、荧光原位杂交的探针 三、荧光原位杂交探针的制备和荧光原位杂交(按试验流程介绍) 一、概述 1、克隆性染色体异常是肿瘤的特征 1914年德国遗传学家Boveri就提出染色体畸变与肿瘤起源相关,然而这还仅仅只是一个假说;1960年Nowell和Hungerford在7例慢性髓系白血病(chronic myeloid leukemia,CML)的患者中发现后来被称为费城染色体(Philadelphia chromosome)的微小染色体;1973年Rowley证实了Ph染色体是9号和22号染色体易位所致,这是人们在肿瘤中认识到的第一个染色体易位;目前,已经有11,500篇文献报道了55,600多种克隆性细胞遗传学异常。这些染色体畸变,尤其是染色体易位及其相应的融合基因在肿瘤致病的起始阶段有着重要的作用,无不说明克隆性细胞遗传学异常是肿瘤的特征,在肿瘤起源中起重要作用。
下图是各种疾病报告的克隆性染色体异常病例数
2、染色体异常的常见类型 染色体异常指数目异常和结构异常两类:前者包括整条染色体数目的扩增和缺失;后者包括染色体易位、插入、倒置、区带的缺失或扩增等。 下图是染色体数目异常
染色体结构异常 3、染色体异常的检测方法 染色体异常的识别得益于二十世纪六十年代后发展起来的胰蛋白酶-姬姆萨染色和常规显带技术,使得常规筛查全基因组染色体异常和检测染色体核型改变成为可能。染色体显带是细胞遗传学分析技术中标准和常用的方法,但耗时且依赖于获得良好的分裂相,还难于分析复杂和隐匿的异常。
qPCR引物设计原则及具体操作步骤 1.找基因(DNA) 1)通过英文名称查找 通过查看文献或者百度搜索查找到对应基因的准确的英文名称 →进入NCBI官网 →点击网页右下角GenBank,进入GenBank界面 →在搜索框中输入准确的英文名称,点击Search搜索即可 2)通过序列号查找 通过查找文献,找到相应基因在GenBank上的登录号,直接输入上面的搜索框进行查找即可。 例如:犬冠状病毒(canine coronavirus,CCV)基因保守片段序列号为KT222978。 3)通过引物查找 通过查找文献,找到别人用过的对应的引物 →在NCBI官网右下角点击Primer-BLAST →输入正、反向引物序列 →设置对应参数 →点击“Get Primers”进行搜索即可 4)找到对应的基因后点击“FASTA”,进入相应界面,再点击“Send to”选择相应格式,保存 序列。
2.qPCR引物和TaqMan探针的设计 1)引物设计注意事项 a)引物长度17bp-25bp为佳。太短的引物容易导致扩增效率降低;太长的引物会导致出 现引物高级结构的几率增加。两者都会干扰定量结果的准确性 b)扩增片段长度为:90-150 bp(最低不能超过70,最高不能超过180) c)引物的Tm值为:最小57℃,最大63℃,最适为60℃,两条引物之间退火温度得差距 不超过1℃,推荐使用Primer Premier 5进行Tm值计算; d)引物A、G、C、T整体分布尽量要均匀,避免使用GC或者TA含量高的区域,尤其 是3’端,必须避开GC含量不均匀的区域。 e)引物设计时请尽量避开TC或者AG的连续结构。 f)3’端不能超过3个以上碱基互补,自互补碱基数不超过3;3’端最后一个碱基绝对不能 搭上 g)特异性要有保证,与非特异模板3’端互搭碱基数不超过3,不连续出现4个及以上的 GC互搭 h)引物3’端最后五个碱基不能包含超过2个以上的G或者C i)引物的GC含量控制在40%-60%之间为好,最佳为45%-55%之间 j)正向或者反向引物应尽量接近探针序列但是不能和探针序列有重合区域 k)在Primer-BLAST设计时,在Organism 处选择相应物种 l)需跨外显子设计,避免基因组污染 2)TaqMan探针设计指南 a)探针序列应尽量接近正向或者反向引物,但是不能与之有重合区域;一般相隔1~5个 碱基(一般10个以内,最好是1个碱基)。 b)应避免连续相同的碱基出现,特别是要避免GGGG或者更多的连续G出现。 c)探针5’端应避免使用碱基G,因为5'G会有淬灭作用,而且即使是被切割下来还会存 在淬灭作用 d)3’端应避免使用碱基A
本文叙述了一种用于甲基化分析的探针法定量PCR的引物和探针设计方法,目前用于甲基化检测的引物探针设计工具非常多,都有使用成功的案例,经过初步多方尝试,本文中叙述的为本人认为较为靠谱的方法。Oligo7的优势在于专业,参数详尽且可自由设置,模块化设计,学会后使用便利。专业的活就是要专业的用专业的工具干。
首先是进行序列转换,有较多的在线工具和联机软件都可实现,这里使用https://www.doczj.com/doc/da8649244.html,/methprimer/,较为简单直观。
直接将目标序列放入如上图的编辑框中,此也可直接用于相关引物的设计,不过本人没使用过,因为不能设计探针。submit后就有转化后的序列信息,如下图: 以上详细标记了CpG位置和非CpG位置的C,可直接复制到Word标注使用,下面就可以使用Oligo7利用上边的序列设计引物和探针了,如果是设计非甲基化引物探针,则使用原始序列。
关于引物和探针的一些主要参数,主要参考invtrogen的建议: Primer设计的基本原则: a)引物长度一般在18-35mer。 b)G-C含量控制在40-60%左右。 c)避免近3’端有酶切位点或发夹结构。 d)如果可能避免在3’端最后5个碱基有2个以上的G或C。 e)如果可能避免在3’端最后1个碱基为A。 f)避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。 g)退火温度Tm控制在58-60C左右。 h)如果是设计点突变引物,突变点应尽可能在引物的中间。 T aqMan 探针设计的基本原则: a)T aqMan 探针位置尽可能靠近扩增引物(扩增产物50-150bp),但不能与引物重叠。 b)长度一般为18-40mer 。 c)G-C含量控制在40-80%左右。 d)避免连续相同碱基的出现,特别是要避免GGGG或更多G出现。 e)在引物的5’端避免使用G。 f)选用比较多的碱基C。 g)退火温度Tm控制在68-70℃左右。 另:目标变异碱基最好在3’末端或3’末端-1位置,保证扩增特异性,对于甲基化,则最好是C。
原位杂交 (In situ hybridization) 一、目的 掌握核酸探针原位杂交操作技术,并利用该技术对单细胞的靶目标进行定位,用于细胞生物学基础研究。 二、原理 原位杂交技术(in situ hybridization)是分子生物学和组织化学成功结合的产物,是特定标记的已知序列核酸作为探针与细胞或组织切片中核酸进行杂交并对其实行检测的方法。其基本原理是含互补序列的标记DNA或RNA片段,即探针,在适宜的条件下与细胞内特定的DNA或RNA形成稳定的杂交体。 原位杂交能在成分复杂的组织中进行单一细胞的研究而不受同一组织中其它成分的影响,因此对于那些细胞数量少且散在于其他组织中的细胞内DNA或RNA研究更为方便;由于原位杂交不需要从组织中提取核酸,对于组织中含量极低的靶序列有极高的敏感性,并可完整地保持组织与细胞的形态,更能准确地反映出组织细胞的相互关系及功能状态。 三、仪器设备 烘箱,切片机,展片机,染色缸,湿盒,原位PCR仪,显微镜、镊子、量筒、烧杯、吸水纸、枪与枪头、载玻片、盖玻片、冰盒等。 四、材料和试剂 1.材料:带有病原体的水生动物,如感染WSSV病毒的对虾、感染虹彩病毒的水生动物等。 2.试剂: Davidson’s AFA固定液: 330 ml 95%乙醇 220 ml 福尔马林(37~39%甲醛水溶液) 115 ml 冰醋酸 335 ml H2O 混匀后封口,室温放置; DIG标记与检测试剂盒(Roche公司); TNE:50 mmol/L Tris-HCl 6.57 g Tris Base 10 mmol/L NaCl 0.58 g NaCl 1 mmol/L EDTA 0.37 g EDTA ddH2O 900 ml (定容至1L) 用HCl调pH至7.4,高压灭菌,4℃保存;
两种定量分析方法的比较及Taqman 探针、引物设计原则 遗传物质DNA 首先要把所携带的遗传信息转录成为信使RNA (mRNA ),携带遗传信息的mRNA 从细胞核进入到细胞质中与核糖体结合,在核糖体中mRNA 携带的遗传信息被翻译成为多肽,多肽经过进一步加工后变成蛋白质,至此遗传物质DNA 完成了表达过程。期间的转录过程是基因表达中非常重要的调节步骤,所转录的mRNA 的多少直接影响着相关最终蛋白质的多少,所以通过对细胞内某条基因mRNA 含量多少的分析,就能大致判断出该条基因的表达是否活跃。 定量PCR 仪是在普通PCR 仪的基础上加装了荧光激发装臵和荧光检测装臵,PCR 扩增和检测同时进行;在PCR 反应体系中加入荧光基团,利用荧光信号的积累实时监测整个PCR 进程,最后通过标准曲线对未知模板进行定量分析。该技术于1996年由美国Applied Biosystems 公司推出,由于该技术不仅实现了PCR 从定性到定量的飞跃,而且与常规PCR 相比,它具有特异性更强、有效解决PCR 污染问题、自动化程度高等特点,目前已得到广泛应用。 定量PCR 常用的三个常用概念 扩增曲线、荧光阈值、Ct 值 扩增曲线:反映PCR 循环次数和荧光强度的曲线,定量PCR 仪每次轮PCR 扩增都会自动记录 荧光强度的变化 荧光阈值:样本的荧光背景值和阴性对照的荧光值,手动 设臵的原则要大于样本的荧光背 景值和阴性对照的荧光最高值,同时要尽量选择进入指数期的最初阶段,并且保 证回归系数大于0.99。 CT 值: PCR 扩增过程中,扩增产物的荧光信号达到设定的阈值时所经过的扩增循环次数。 扩增曲线 阈值及CT 值 荧光定量PCR 的数学原理 理想的PCR 反应: X=X0*2n 非理想的PCR 反应: X=X0* (1+Ex)n (n :扩增反应的循环次数;X :第n 次循环后的产物量;X0:初始模板量;Ex :扩增效率) 在扩增产物达到阈值线时 : C(t) value
实时荧光Taqman 探针设计的几个要点 实验室很多同学都要做Real time PCR实验,实验室的师兄师姐都会有很多宝贵意见,不过也有实验室前没有做过的,查找了下资料和大家分享下关于实时荧光Taqman探针设计、实时荧光PCR探针的选择、 引物的设计及评价。 荧光探针法是用序列特异的荧光标记探针来检测产物,探针法的出现使得定量PCR技术的特异性比常规PCR技术大大提高。目前较常提及的有TaqMan探针、FRET杂交探针(荧光共振能量传递探针)和分子信 标Molecular Beacon。 广泛使用的TaqMan探针法是指PCR扩增时在加入一对引物的同时另外加入一个特异性的荧光探针,该探针只与模板特异性地结合,其结合位点在两条引物之间。探针的5′端标记有荧光报告基团(Reporter, R),如FAM、VIC等,3′端标记有荧光淬灭基团(Quencher, Q),如TAMRA等。当探针完整的时候,5′端报告基团经仪器光源激发的荧光正好被近距离的3′端荧光基团淬灭,仪器检测不到5′端报告基团所激发的荧光信号(就是说5’荧光基团的发射波长正好是3’ 荧光基团的吸收波长,因而能量被吸收传递到3’荧光基团而发出其它荧光)。随着PCR的进行,Taq酶在链延伸过程中遇到与模板结合的探针,其5′-3′外切酶活性(此活性是双链特异性的,游离的单链探针不受影响)就会将切割探针,释放5′端报告基团游离于反应体系中,远离3′端荧光淬灭基团的屏蔽,5′端报告基团受激发所发射的荧光信号就可以被探头检测到。也就是说每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。报告信号 的强度就代表了模板DNA的拷贝数。 (请注意,该图显示的不是普通的Taqman探针法,而是Taqman MGB探针法)Taqman探针检测的是积累荧光。常用的荧光基团有FAM,TET,VIC,HEX等等。当探针完整的时候,由于3′端的荧光淬灭基团在吸收5′端报告基团所发射的荧光能量,本身会发射波长不同的荧光而导致本底高,因此TaqMan探针近来又有新的发展——TaqMan MGB探针。MGB探针的淬灭基团采用非荧光淬灭基团(Non-Fluorescent Quencher),本身不产生荧光,可以大大降低本底信号的强度。同时探针上还连接有MGB (Minor Groove Binder)修饰基团,可以将探针的Tm值提高10°C左右。因此为了获得同样的Tm值,MGB探针可以比普通TaqMan探针设计得更短,既降低了合成成本,也使得探针设计的成功率大为提高——因为在模板的DNA碱基组成不理想的情况下,短的探针比长的更容易设计。实验证明,TaqMan MGB探针对于富含A/T 的模板可以区分得更为理想。 Taqman探针法已经得到广泛使用,不过有人认为这种技术利用了Taq酶5`—3`外切酶活性,一般试剂厂家只给Taq酶的聚合酶活性定标,没有同时给Taq酶5`—3`外切酶活性定标,不同批号试剂之间会给定量带来差异。另外对探针的熔点温度(Tm)仅要求其高于60°C,这就使不同试剂盒之间的特异性参差不齐,难 于做质控检测。 Real time PCR Taqman探针设计、实时多重PCR探针的选择和引物的设计及评价 一、实时荧光Taqman探针设计 总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。即real-time PCR的扩增片段是50bp----150bp。当找不到150bp的保守片段时,必须确保探针的 片段是保守的。
原位杂交实验原理与方法 一、目的 本实验的目的是学会原位杂交的使用方法。了解各种原位杂交的基本原理和优缺点。 二、原理 原位杂交组化(简称原位杂交,in situ hybridization histochemistry;ISHH)属于分子杂交的一种,是一种应用标记探针与组织细胞中的待测核酸杂交,再应用标记物相关的检测系统,在核酸原有的位置将其显示出来的一种检测技术。原位杂交的本质就是在一定的温度和离子浓度下,使具有特异序列的单链探针通过碱基互补规则与组织细胞内待测的核酸复性结合而使得组织细胞中的特异性核酸得到定位,并通过探针上所标记的检测系统将其在核酸的原有位置上显示出来。 当然杂交分子的形成并不要求两条单链的碱基顺序完全互补,所以不同来源的核酸单链只要彼此之间有一定程度的互补顺序(即某种程度的同源性)就可以形成杂交双链。 探针的种类按所带标记物可分为同位素标记探针和非同位素标记探针两大类。目前,大多数放射性标记法是通过酶促反应将标记的基因掺入DNA中,常用的同位素标记物有3H、35S、125I和32P。同位素标记物虽然有灵敏性高,背底较为清晰等优点,但是由于放射性同位素对人和环境均会造成伤害,近来有被非同位素取代的趋势。非同位素标记物中目前最常用的有生物素、地高辛和荧光素三种。 探针的种类按核酸性质不同又可分为DNA探针、cDNA探针、cRNA探针和合成寡核苷酸探针。cDNA探针又可分为双链cDNA探针和单链cDNA探针。 原位杂交又可分为菌落原位杂交和组织原位杂交。 菌落原位杂交(Colony in situ hybridization)菌落原位杂交是将细菌从培养平板转移到硝酸纤维素滤膜上,然后将滤膜上的菌落裂菌以释出DNA。将NDA烘干固定于膜上与32P 标记的探针杂交,放射自显影检测菌落杂交信号,并与平板上的菌落对位。 组织原位杂交(Tissue in situ hybridization)组织原位杂交简称原位杂交,指组织或细胞的原位杂交,它与菌落的原位杂交不同。菌落原位杂交需裂解细菌释出DNA,然后进行杂交。而原位杂交是经适当处理后,使细胞通透性增加,让探针进入细胞内与DNA或RNA 杂交。 (一)探针的选择 根据不同的杂交实验要求,应选择不同的核酸探针。在大多数情况下,可以选择克隆的DNA 或cDNA双链探针。但是在有些情况下,必须选用其它类型的探针如寡核苷酸探针和RNA探针。例如,在检测靶序列上的单个碱基改变时应选用寡核苷酸探针,在检测单链靶序列时应选用与其互补的DNA单链探针(通过克隆人M13噬菌体DNA获得)或RNA探针,寡核苷酸探针也可。长的双链DNA探针特异性较强,适宜检测复杂的靶核苷酸序列和病原体,但不适宜于组织原位杂交,因为它不易透过细胞膜进入胞内或核内。在这种情况下,寡核苷酸探针和短的PCR标记探针(80~150bp)具有较大的优越性。 在选用探针时经常会受到可利用探针种类的限制。如在建立DNA文库时,手头没有筛选特定基因的克隆探针,这时就可用寡核苷酸探针来代替。但必须首先纯化该基因的编码蛋白,并测定6个以上的末端氨基酸序列,通过反推的核苷酸序列合成一套寡核苷酸探针。如果已有其它动物的同种基因克隆,因为人类和动物间在同一基因的核苷酸顺序上存在较高的同源性,因此可利用已鉴定的动物基因作探针来筛选人类基因克隆。对于基因核苷酸序列背景清楚而无法获得克隆探针时,可采用PCR方法扩增某段基因序列,并克隆人合适的质粒载体中,
原位杂交技术的操作详解及小贴士 原位杂交技术应用于染色体、细胞和组织切片等样品中进行核酸特异性检测,与免疫组化技术的结合应用,能将DNA、mRNA和蛋白水平上的基因活性与样品的显微拓扑信息结合起来。1969年Pardue和Gall将放射性标记的探针直接应用于纯化核酸的杂交,此后得益于分子克隆技术的发展,及不同探针标记系统和检测系统的应用,大大增加了原位杂交检测的应用灵活性和检测灵敏度。 多种探针标记检测系统 基于地高辛、生物素和荧光标记分子的标记和检测系统是常见的原位杂交检测方法。 荧光标记检测常为直接探针标记方法,如在dUTP/UTP/ddUTP上连接Fluorescein后进行核酸标记。由于标记在核酸上的荧光分子必须经受杂交和洗脱过程中的考验,以及荧光分子易于衰减,其检测灵敏度受到一定的影响。但对荧光分子的直接检测呈现的背景较低。 间接标记的方法中应用了报告分子标记的探针,报告分子通过亲和酶促的方法进行显色。常用的报告分子如地高辛,生物素。结合地高辛抗体或链霉亲和素上耦联的酶系统进行间接的底物反应检测。地高辛标记核酸的历史可追溯到1987年,由于地高辛是洋地黄的花和叶中特有的成分,检测时使用的地高辛抗体不会结合于其他的生物分子。这是相较于生物素标记系统的优势。地高辛抗体上可耦联碱性磷酸酶、过氧化酶,及荧光分子和胶体金等,根据不同的应用需求,呈现高信噪比的核酸检测结果。但需注意,由于引入了免疫检测反应,在放大检测灵敏度的同时,应注意样品内源性酶的灭活,以降低检测背景。 通过不同标记方法的联合应用,还可在同一样本中实现染色体不同区域或细
胞样本中不同RNA序列的多重检测。 原位杂交中探针的选择 DNA探针、RNA探针和寡核苷酸探针均能通过不同的酶促分子反应进行标记。寡核苷酸探针的长度较短,因此避免了探针内部退火的问题,在杂交时的渗透能力也更好,探针与靶标的接触这是影响原位杂交是否成功的重要因素之一。DNA 探针、RNA探针在合成时需要控制探针片段长度,通常300-1000bp左右,能覆盖到较长片段的靶核酸序列,增加检测的灵敏度。 就DNA探针和RNA探针的比较,DNA探针在杂交过程中会出现探针双链之间退火的可能,也更倾向于在溶液中形成大分子的探针聚合体,从而影响其渗透能力。而RNA 探针的应用,将提高DNA-RNA杂交子的热稳定性。 Tips:RNA探针因其单链、高分子结合力、可适应高温杂交的特性,其检测特异性和灵敏度均优于DNA探针。常用的RNA探针标记方法为构建质粒后进行转率合成。通过PCR扩增的方法,可以更方便地进行RNA探针的制备;RNA探针合成后,还需验证其对目标片段检测的灵敏度和特异性。具体实验流程和注意事项可参考技术文章:A Method for High Quality Digoxigenin-Labeled RNA Probes for In Situ Hybridization 原位杂交检测步骤 原位杂交涉及的步骤:玻片的准备和样品固定,细胞或组织的预渗透处理,靶DNA变性(DNA原位杂交),探针制备,原位杂交过程,杂交后洗涤,探针(显色)检测。 1. 玻片的准备和样品固定 对于染色体涂片,1:1的乙醇/醚处理的载玻片已能符合要求。对于组织切片的原位杂交,为了在实验过程中不丢失组织样品,可使用多聚赖氨酸或铬矾
两种定量分析方法的比较及Taqman探针、引物设计原则 遗传物质DNA首先要把所携带的遗传信息转录成为信使RNA(mRNA),携带遗传信息的mRNA从细胞核进入到细胞质中与核糖体结合,在核糖体中mRNA携带的遗传信息被翻译成为多肽,多肽经过进一步加工后变成蛋白质,至此遗传物质DNA完成了表达过程。期间的转录过程是基因表达中非常重要的调节步骤,所转录的mRNA的多少直接影响着相关最终蛋白质的多少,所以通过对细胞内某条基因mRNA含量多少的分析,就能大致判断出该条基因的表达是否活跃。 定量PCR仪是在普通PCR仪的基础上加装了荧光激发装置和荧光检测装置,PCR扩增和检测同时进行;在PCR反应体系中加入荧光基团,利用荧光信号的积累实时监测整个PCR 进程,最后通过标准曲线对未知模板进行定量分析。该技术于1996年由美国Applied Biosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR 相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。 定量PCR常用的三个常用概念 扩增曲线、荧光阈值、Ct值 扩增曲线:反映PCR循环次数和荧光强度的曲线,定量PCR仪每次轮PCR扩增都会自动记录荧光强度的变化 荧光阈值:样本的荧光背景值和阴性对照的荧光值,手动设置的原则要大于样本的荧光背景值和阴性对照的荧光最高值,同时要尽量选择进入指数期的最初阶段,并且保 证回归系数大于0.99。 CT值: PCR扩增过程中,扩增产物的荧光信号达到设定的阈值时所经过的扩增循环次数。 C(t) value 扩增曲线阈值及CT值 荧光定量PCR 的数学原理