大疱性表皮松解详解
- 格式:pdf
- 大小:2.70 MB
- 文档页数:15

大疱性表皮松解有哪些症状?*导读:本文向您详细介绍大疱性表皮松解症状,尤其是大疱性表皮松解的早期症状,大疱性表皮松解有什么表现?得了大疱性表皮松解会怎样?以及大疱性表皮松解有哪些并发病症,大疱性表皮松解还会引起哪些疾病等方面内容。
……*大疱性表皮松解常见症状:肠闭锁、结痂、疱疹、大疱(含脓性液体)*一、症状1.单纯型大疱性表皮松解症(EBS)是以表皮内水疱为特征,主要由角蛋白突变所引起的一组遗传性皮肤病,侵袭1/4万人群,根据临床的严重性进一步分成不同亚型,单纯型大疱性表皮松解症家族的外显率高,且它最严重的亚型,疾病在出生时就表现明显。
至少有11种亚型的单纯型大疱性表皮松解症,其中7种为常染色体显性遗传,3种最常见亚型均为常染色体显性遗传,包括泛发性大疱性表皮松解症(Koebnet),局限性大疱性表皮松解症(Weber Cockayne)和疱疹样大疱性表皮松解症(Dowling Meata,表2),随着年龄增长起疱可显著减少,有时可几个月不起疱,可能是随着患者年龄长大,表皮充分伸展,其所受的机械性张力自然减小。
(1)泛发性大疱性表皮松解症:始于新生儿至婴儿早期,多见于手,足和四肢,也可见掌跖过度角化和脱屑,多不累及甲,齿和口腔黏膜。
(2)局限性大疱性表皮松解症:始于儿童时期或更晚,是最常见的一型,也可以到成人时才出现,表现为在高强度运动后出现手,足部厚壁水疱,常见手足多汗,足部的水疱常继发感染。
(3)疱疹样大疱性表皮松解症:出生时即可见,是最严重的一型,水疱泛发全身,可累及口腔黏膜,婴儿期可出现明显的炎症伴粟粒疹,在儿童早期水疱多不结痂,躯干部和四肢近端可白发成群或“疱疹样”水疱,因为水疱裂隙位于表皮内,愈后不留瘢痕,指(趾)甲可能脱失,但通常可再生。
与前两型不同,遇热后水疱不会加重,到6,7岁时可出现掌跖角化过度,尽管一些患者水疱非常严重,但很少危及生命,因为局限性皮肤屏障功能丧失,易于继发感染。

大疱性表皮松解疾病研究报告疾病别名:大疱性表皮松解所属部位:皮肤就诊科室:病症体征:败血症,大疱(含脓性液体),疱疹,鳞状细胞癌,结痂疾病介绍:什么是大疱性表皮松解?大疱性表皮松解是怎么回事?大疱性表皮松解(EPIDERMOLYSIS BULLOSA,EB)由KOEBNER在19世纪晚期首次提出,用以描绘一种不留瘢痕的水疱性皮肤病,随后用于描述一组以皮肤和黏膜对机械损伤易感并形成大疱为特征的多基因遗传性皮肤病,为一组典型的侵及皮肤基底膜区的疾病,内脏器官也可累及,临床上病情表现出极大的变异性,同时,基因杂合性也很明显,有常染色体显性和隐性遗传,异常的伤口修复可导致慢性损害和结痂,转移性癌也常见,目前,对本病的研究取得显著进展,其研究手段主要是通过分子克隆编码一些维持皮肤层次结构完整的关键蛋白基网,本病亦属于中医的天疱疮范畴症状体征:大疱性表皮松解有什么症状?以下就是关于大疱性表皮松解有哪些症状的详细介绍:1.单纯型大疱性表皮松解症(EBS)是以表皮内水疱为特征,主要由角蛋白突变所引起的一组遗传性皮肤病,侵袭1/4万人群,根据临床的严重性进一步分成不同亚型,单纯型大疱性表皮松解症家族的外显率高,且它最严重的亚型,疾病在出生时就表现明显。
至少有11种亚型的单纯型大疱性表皮松解症,其中7种为常染色体显性遗传,3种最常见亚型均为常染色体显性遗传,包括泛发性大疱性表皮松解症(KOEBNET),局限性大疱性表皮松解症(WEBER COCKAYNE)和疱疹样大疱性表皮松解症(DOWLING MEATA,表2),随着年龄增长起疱可显著减少,有时可几个月不起疱,可能是随着患者年龄长大,表皮充分伸展,其所受的机械性张力自然减小。
(1)泛发性大疱性表皮松解症:始于新生儿至婴儿早期,多见于手,足和四肢,也可见掌跖过度角化和脱屑,多不累及甲,齿和口腔黏膜。
(2)局限性大疱性表皮松解症:始于儿童时期或更晚,是最常见的一型,也可以到成人时才出现,表现为在高强度运动后出现手,足部厚壁水疱,常见手足多汗,足部的水疱常继发感染。



大疱性表皮松懈症大疱性表皮松解症是一组慢性非炎症性大疱性疾病,其特点是皮肤黏膜在轻微外伤后自然发生大疱,基本病理改变是水疱位于真皮或基底膜,但有些原发缺陷位于表皮。
类型与诊治编辑本段回目录大疱性表皮松解症的基本类型根据临床表现、遗传方式和电镜检查的特点,可分成下列6型.(一)单纯型大疱性表皮松解症(Epidermolysis bullosa simplex)【临床表现】本病是表皮对机械性损伤的反应加剧所致,故在易受外伤或压迫的手、足、肘及发生透亮、紧张的水疱或大疱,愈后无疤痕,黏膜及指甲很少累及。
本病损害数目少,常在出生后头几年发病,青春期后常减轻消退。
【诊断及鉴别诊断】主要根据1岁左右发病,大疱位于手足、肘和膝等部位确定诊断。
【治疗】避免外伤及受热,在4个月至2岁间尤为重要,要避免穿质地粗糙的衣服,以防止摩擦发生水疱和大疱。
一旦发生要注意护理,防止感染。
足跖部水疱应用消毒空针抽吸疱液后包扎。
严重病例可用糖皮质激素,有人认为大剂量维生素E或枸橼酸钠口服对本病有效,后者用量3次/d,每次2g。
有人用四环素500mg,3次/d,有效。
可试用美满霉素。
(二)Coekayne手足大疱性表皮松解症(Epidermolysis bullosa of the hands and feet Cockayne)【临床表现】起病于儿童。
大疱仅发于手足,尤多见于两足,愈后无疤痕,夏季发病或病情加重。
【诊断及鉴别诊断】根据手足部大疱、起始于儿童确定诊断。
【治疗】避免外伤。
维生素E治疗有效。
上述疗法亦可试用。
(三)显性遗传性营养障碍性大疱性表皮松解症(Epidermolysis bullosa dystrophica dominent)又名增生性大疱性表皮松解症。
【临床表现】起病于幼婴及儿童,亦可较迟。
水疱及大疱位于四肢伸侧,尤以关节部位多见,多由外伤引起,愈后遗留萎缩性疤痕,亦可为肥大性疤痕及疤痕疙瘩,指、趾甲常缺损。


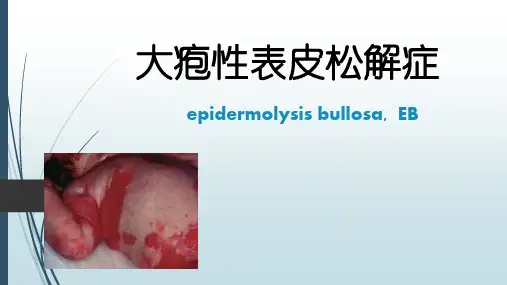

后天性大疱性表皮松解是怎么回事?
*导读:本文向您详细介绍后天性大疱性表皮松解的病理病因,后天性大疱性表皮松解主要是由什么原因引起的。
*一、后天性大疱性表皮松解病因
*一、发病原因
病因尚不清楚,但有相当多的证据表明本病的发生与自身免疫因素有关。
直接免疫荧光检查发现在皮损旁皮肤活检真表皮结合部位可见IgG沉积。
*二、发病机制
最近的研究表明这种自身抗体是抗位于锚原纤维的Ⅶ型胶原蛋白。
这种IgG抗体可与Ⅶ型胶原的a链结合,导致锚原纤维数量减少。
这种减少的机制尚不清楚,有人认为,可能新合成Ⅶ型胶原纤维的a链与EBA自身抗体结合,使得三角螺旋结构和稳定锚原纤维不能形成所致。
体外实验尚未能证实EBA自身抗体IgG能引起皮肤水疱形成。
获得性大疱性表皮松解病虽不是遗传性疾病,但确实有自身免疫的遗传素质,在美国的东南部黑人中,EBA和大疱性SLE患者均有高的HLA-DR2表型阳性率,这一结果也提示EBA和大疱性SLE在免疫遗传学上是有关联的,HLA-DR2基因参与抗锚原纤维
胶原蛋白的自身免疫反应。
*温馨提示:以上就是对于后天性大疱性表皮松解病因,后天性大疱性表皮松解是由什么原因引起的相关内容叙述,更多有关后天性大疱性表皮松解方面的知识,请继续关注疾病库,或者在站内搜索“后天性大疱性表皮松解”找到更多扩展内容,希望以上内容可以帮助到您!。

可治性罕见病—遗传性大疱性表皮松解症一、疾病概述遗传性大疱性表皮松解症(epidermolysis bullosa,EB)是一种遗传性皮肤病,呈常染色体显性或隐性模式遗传。
表现为皮肤脆性增加,皮肤黏膜受轻微外力摩擦后起水疱、大疱样皮疹,可伴表皮剥脱和黏膜损害等。
患儿出生后不久即出现皮肤损伤,主要见于易受压部位,水疱逐渐扩大且容易破溃,护理不当易继发细菌感染,皮损反复发作,可致瘢痕形成。
超微结构研究证明其基本病理改变是位于表皮真皮交界处的各种连接蛋白缺陷所致,基因突变在本病的发病中起重要作用。
1988年,国际上首次召开了有关EB诊断和分类的研讨会并提出了最初的分类意见[1],临床上将EB分为3型:单纯型(epidermolysis bullosa simplex,EBS)、交界型(junctional epidermolysis bullosa,JEB)和营养不良型(dystrophic epidermolysis bullosa,DEB)。
单纯性先天性大疱性表皮松解症裂隙位于表皮基底角质形成细胞,与编码角蛋白5和14及编码网格蛋白的PLEC1的基因突变有关。
交界性大疱性表皮松解症裂隙位于基膜透明板,与本病有关的基因主要有:LAMB3、COL1 7A1、LAMC2和LAMA3,这些基因的突变导致皮肤黏膜变脆,板层素5(laminin5)、α6,β4-整合素和大疱性类天疱疮抗原2(BPAG2)的基因表达异常也可以导致本病。
营养不良型裂隙位于基膜致密板下带,是因Ⅶ型胶原缺少所致,最终导致致密板的固定结构锚纤维减少而发病。
本病的诊断靠基因定位、透射电子显微镜和免疫荧光检测抗原或抗体等检查综合分析,透射电子显微镜是诊断本病最有效的方法。
2007年,第三次研讨会在最新的分子遗传学研究基础上对以往的认识进行完善和修正,加入了一些新近明确的病种或亚型,也除外或重新命名了一些以前认识上有偏差的疾病,这一新的分类标准已于2008年正式发布[2]。

皮肤科大疱性表皮松解症的诊疗护理大疱性表皮松解症(epidermo1ysisbu11osa,EB),分遗传性和获得性两类。
遗传性大疱性表皮松解症是一组临床上以轻微外伤后皮肤和黏膜水疱形成为共同特征的基因遗传性皮肤病,皮肤脆性增加,常伴有皮肤外表现,临床表现依分型不同轻重不O【病因及发病机制】所有类型的大疱性表皮松解症,其特征性的表现为皮肤对机械力脆性增加。
原因涉及十多个编码结构蛋白中的基因突变,这些蛋白质通常位于表皮、真表皮交界处或者真皮乳头上层。
发生突变的蛋白质在皮肤的分布决定大疱性表皮松解症大疱的位置。
绝大多数单纯型大疱性表皮松解症都是常染色体遗传。
所有类型的交界型大疱性表皮松解症都是常染色体隐性遗传,从父母各遗传了一个突变单体,父母是携带致病基因的健康人,每一次怀孕都有1/4的可能性使孩子患病。
而每个孩子都有1/2的概率成为像其父母一样的健康携带者。
营养不良型EB为常染色体显性遗传或常染色体隐性遗传。
【临床类型及表现】1临床类型遗传性大疱性表皮松解症分为单纯型大疱性表皮松解症(EBS).交界型大疱性表皮松解症(JEB).营养不良型大疱性表皮松解症(DEB)及Kind1er综合征。
2.临床表现机械性脆性皮肤、张力性大疱和糜烂结痂是各型遗传性大疱性表皮松解症共有的特点。
瘢痕(几乎都是萎缩性)可发生于任何类型和亚型的大疱性表皮松解症,包括单纯型大疱性表皮松解症。
皮肤外的表现:大疱性表皮松解症患者皮肤的分子缺陷同样会出现在其他存在上皮的组织,包括眼、口腔、消化道和泌尿生殖道。
当眼部受累时,可导致新生血管形成和失明。
若食管慢性持续性受累,则可形成瘢痕、缩窄甚至完全堵塞。
小肠受累引起慢性营养吸收不良,大肠受累少见,但也可导致严重的便秘、肛裂和肛门狭窄。
严重的患者还可出现食管反流。
所有交界型大疱性表皮松解症患者都有牙釉质发育不良,乳牙和恒牙表面有点状缺损。
隐性遗传性营养不良型大疱性表皮松解症(RDEB)患者可发生假性并指,最初表现为近端的指(趾)噗融合,如未予治疗,继续发展,指(趾)就会被瘢痕组织包绕形成永久性的并指(趾)。