Gitelman综合征
- 格式:docx
- 大小:123.85 KB
- 文档页数:6



2023,26(1):116-119.[10]㊀Guo Z ,Zhang K ,Wei X ,et al.Radiotherapy plus camreli-zumab affects peripheral CD8T -cell differentiation subsetsexpressing PD -1,TIGIT ,and CTLA -4in esophageal squa-mous cell carcinoma [J ].Leukoc Biol ,2023,113(1):11-17.文献综述ʌ文章编号ɔ1006-6233(2023)11-1933-04Gitelman 综合征的发病机制和临床表现相关研究进展金更学,㊀徐翔宇,㊀陈㊀慧(兰州大学第二医院内分泌代谢科,㊀甘肃㊀兰州㊀730030)ʌ关键词ɔ㊀Gitelman 综合征;㊀低钾血症;㊀基因突变ʌ文献标识码ɔ㊀A㊀㊀㊀㊀㊀ʌdoi ɔ10.3969/j.issn.1006-6233.2023.11.034㊀㊀Gitelman 综合征(Gitelman syndrome ,GS )亦称家族性低钾低镁血症,由Gitelman 于1966年首次提出,是一种常染色体隐性失盐性肾小管遗传病㊂据估计,在国外GS 的患病率约为1~10/40000,其中杂合子约占1%左右,亚洲地区可能存在更高的患病率,但目前国内尚无相关统计数据㊂2018年5月11日,中国国家卫生健康委员会等部门联合制定了‘第一批罕见病目录“,将GS 列入其中㊂该综合征生化筛查重点为肾性失钾㊁低钾血症㊁代谢性碱中毒㊁低钙尿症㊁低镁血症和高肾素血症性醛固酮增多症㊂具有起病隐匿,临床表现不典型的特点,主要由低血钾和低血镁引发严重乏力㊁纳差㊁抽搐以及夜尿增多等症状㊂病因为由于基因突变导致肾远曲小管噻嗪敏感的钠氯共转运蛋白(sodium -chloride cotransporter ,NCC )功能丧失,重吸收钠离子和氯离子障碍㊂GS 不仅对患者的日常生活和工作产生不良影响,还容易合并糖代谢异常㊁生长发育与骨关节改变㊁肾脏疾病(远端肾小管性酸中毒㊁肾功能不全)㊁高血压㊁自身免疫性甲状腺疾病㊁原发性醛固酮增多症㊁干燥综合征㊁横纹肌溶解㊁癫痫,甚至心律失常等[1]㊂然而这些症状缺乏特异性,诊断依据以临床表现结合基因检测结果为主,加之基因检测难度和费用较高,漏诊率高,导致临床医生对该病认识不够深入,临床诊疗难度大㊂本文总结GS 的发病机制和临床表现,旨在提高临床医生对GS 的诊疗水平,早诊断早治疗,以期改善患者生活质量㊂1㊀GS 的致病基因GS 常见的致病基因为SLC12A3基因㊂还有Tr-pm6-Mg ㊁CLCKNB ㊁KCNJ10㊁FXYD2㊁HNF1B 突变可能导致类似的表型,因为这些突变间接降低了NCC 活性[2]㊂此外,Na +耗损小管病患者中也检测到MT -TI ㊁MT -TF ㊁KCNJ16和ATP1A1的致病变异[3]㊂这些新发现强调了细胞代谢和基侧膜电位对肾远曲小管中Na +重吸收的重要性,见图1㊂由于约10%的GS 患者中存在未知基因型,少数严重病例仍以临床诊断为主㊂引起基因变异的原因可能与基因修饰㊁性别㊁基因型㊁补偿机制㊁环境因素和饮食习惯等综合因素相关㊂图1㊀GS 的致病基因图㊃3391㊃ʌ基金项目ɔ兰州大学第二医院萃英科技创新计划项目,(编号:CY2018-ZD02);兰州大学第二医院博士研究生培养专项基金资助项目,(编号:YJS -BD -02)ʌ通讯作者ɔ陈㊀慧㊀㊀SLC12A3基因:SLC12A3基因(溶质载体家族12成员3)(solute carrier family12member3),位于染色体16q13,是指位于16号染色体长臂,与着丝粒相距13个图距单位,长度约为55kb,由26个独立的外显子组成㊂该基因编码位于肾脏远曲小管的NCC,具有重吸收钠和氯离子的功能㊂NCC是一个1030个氨基酸组成的整合膜蛋白,包含12个跨膜结构域和细胞内外侧的C-和N-末端结构域㊂目前已发现有350多种突变类型,变异类型主要为点变异㊂在中国GS患者中,错义突变占72%以上,并且最常见的是Thr60Met和Asp486Asn[4],然而热点突变仍不清楚㊂Vargas Pous-sou[5]团队对448例病例进行分析表明,在70%的患者中存在两个受影响的等位基因,其中25%为纯合型, 74.9%为复合杂合型,并且还有18%只有一个等位基因发生突变㊂根据导致NCC活性降低或丧失的机制可将SLC12A3突变分为五类:1类突变:合成受损㊂即产生缺失或非活性蛋白质;这些常见于无意义㊁移码㊁剪接位点突变或过早终止密码子㊂2类突变:加工受损;特征是蛋白质完全转录并翻译出来,但检测到错误折叠从而滞留在内质网并被降解㊂3类突变:运输受损;插入细胞膜异常㊂4类突变:功能特性改变;尽管加工和插入细胞膜正常但其功能特性发生了改变㊂5类突变:降解加速㊂Trpm6-Mg基因:Trpm6(瞬时受体电位阳离子通道,M亚家族,成员6)(transient receptor potential cat-ion channel,subfamily M,member6),位于染色体9q21.13,由39个外显子组成㊂该基因编码远曲小管用十二指肠上皮中镁转运蛋白Trpm6-Mg㊂GS由于肾脏和十二指肠中镁转运蛋白的下调而导致低镁血症㊂已报道50余种突变,并在中国报道过1个家系[6]㊂CLCNKb基因:CLCNKb基因(氯化物通道Kb) (chloride channel Kb)㊂主要分布在人体和哺乳动物的耳蜗和肾脏内,分别参与听觉和尿液的形成㊂该基因位于染色体1p36,包括19个外显子,编码电压依赖性氯离子通道蛋白㊂主要分布在髓袢粗升段,CLC-NKb基因突变是Bartter综合征(因此常伴耳聋)的常见致病原因之一,并且少数情况下也出现在GS患者中[7]㊂还有部分GS患者中存在CLCNKb基因和SLC12A3两种基因都突变的情况[8]㊂2㊀GS病理生理学钠㊁氯㊁镁㊁钙和钾离子在肾脏的处理是一个复杂过程,在各种肾小管通道的分子活性影响下进行,其中肾远曲小管具有重要作用㊂在肾脏近端小管大部分被滤过的氯化钠(65%~70%)㊁钙(70%)㊁镁(90%)会被重新吸收㊂滤液到达远曲小管时,剩余的这些离子将通过跨细胞转运机制被重新吸收,使远曲小管成为调节钠㊁氯㊁钙㊁镁重吸收的重要位点,GS患者肾脏病理机制如图2㊂图2㊀GS患者肾远曲小管和集合管病理机制图低钾性碱中毒:SLC12A3基因编码肾远曲小管顶端膜中的NCC通道㊂NCC有助于从管腔中重吸收钠和氯离子㊂SLC12A3基因突变会导致NCC功能丧失,进而使得钠和氯离子输送到集合管中㊂为了重新吸收多余的钠,远曲小管和集合管基侧膜Na+-K+泵表达和近端小管前半段和髓袢升支粗段顶端膜Na+-H+交换增强㊂另外,集合管通过醛固酮介导的顶端膜上皮钠通道表达也会增加㊂这些变化导致小管上皮细胞内钾离子浓度升高,顶端膜对钾离子具有通透性,因此钾离子可顺化学梯度通过肾脏钾通道进入小管液,即钾和氢的分泌,从而导致低钾性碱中毒㊂此外,为增加钾离子的重吸收,集合管闰细胞顶端膜H+-K+交换也会增加,随着钾离子的重吸收氢离子被分泌入小管液最终导致低钾性碱中毒㊂低镁血症:钙和镁的关系是复杂的,目前还没有准确详细的定义㊂在肾远曲小管的顶端膜和十二指肠的镁转运细胞的刷状边界上,存在镁渗透通道㊂在GS 中,镁通道的表达降低㊂远端小管和十二指肠中这些通道的下调会导致尿液和肠道镁的排出增加,从而在GS中出现低镁血症㊂低镁血症可以部分解释GS患者出现胰岛素抵抗,因为镁离子是胰岛素信号通路中的关键因素[9]㊂此外,低镁血症还可能降低焦磷酸酶活性,促进关节中焦磷酸盐的结晶,导致关节疼痛和软骨钙沉着症[10]㊂㊃4391㊃低钙尿症:体内99%的钙以骨盐的形式沉积在骨骼和牙齿中,少量存在于软组织中㊂生化检测血钙是指总钙的检测,而血气检测可以检测游离钙的含量㊂血钙中约有40%与蛋白质结合,5%-10%为可扩散钙,与有机酸(主要是磷酸盐)结合,而50%处于游离状态发挥其活性作用㊂低镁血症可能会导致小肠和骨骼对甲状旁腺激素的敏感性下降㊂这也可能是GS患者对低尿钙缺乏血钙调节反应不足的原因之一㊂相比于正常人群,GS患者的血钙水平较高㊂在一项涵盖304名GS患者的大型欧洲国际横断面研究中,观察到甲状旁腺功能亢进在GS患者中的发病率低(7%),而甲状旁腺功能低下的发病率更高(20%),血全段甲状旁腺激素(intact parathyroid hormone,iPTH)与血清镁呈显著正相关,而iPTH与25羟基维生素D呈负相关[11]㊂中国队列研究发现纯合突变与GS更早发病和更严重的低钙尿症相关[12]㊂肾远曲小管表达的促钙蛋白㊁促镁蛋白与低钙尿症㊁低镁血症相关[13]㊂目前仍需进一步探索低钙尿症的发生机制㊂除此之外,钙异位沉积在软骨㊁腘绳肌和肾小管也有可能是GS患者存在低钙尿症,而没有高钙血症的原因之一㊂肾素-血管紧张素系统活化:流经肾远曲小管起始部致密斑的氯化钠量减少,以及低血容量对入球小动脉牵张感受器的刺激都可能导致肾素血管紧张素系统激活㊂3㊀GS临床表现目前GS可以基本确定致病的突变基因,但其基因型与表型间相关性仍处于不断深入探索阶段㊂主要临床症状特征包括:对盐的渴望(即儿童时期对咸食或咸味食物偏好)㊁肌无力㊁乏力㊁运动障碍或耐力下降㊁晕厥㊁痉挛㊁抽搐㊁感觉异常㊁脚痉挛㊁生长迟缓㊁青春期延迟㊁身材矮小㊁口渴或异常饮酒行为㊁腹部疼痛发作,成年人可能会出现头晕㊁眩晕㊁多尿㊁夜尿㊁关节疼痛㊁心悸和视力问题㊂四分之三的患者有口渴和对盐的渴望增加㊂多数患者喜欢腌制盐水㊁腌黄瓜㊁橙子和柠檬㊂2021年的中国GS诊疗专家共识将中国人群临床表现及生化分级见表1㊁2[14]㊂表1㊀中国GS患者临床表现类别常见(大于50%患者)多见(20~50%患者)偶见(<20%患者)全身症状对盐的渴望㊁疲乏口渴㊁多饮-神经-肌肉系统肌无力手足抽搐㊁肌肉僵硬疼痛㊁感觉异常㊁头晕眩晕㊁共济失调㊁软瘫㊁呼吸困难心血管系统心悸㊁低血压QT间期延长晕厥泌尿系统夜尿增多多尿-骨关节系统-软骨钙化关节痛消化系统--腹痛表2㊀GS患者生化分级建议级别1级2级3级4级血钾(mmoL/l) 3.0~3.4 2.5~2.9 2.0~2.4<2.0,或低钾血症伴轻瘫㊁肠梗阻,或危及生命的心律失常血镁(mmoL/l)0.60~0.700.45~0.590.30~0.44<0.30,或伴危及生命的心律失常,或手足抽搐GS发病人群主要集中在青春期或成人阶段,但也有报道称2岁㊁5岁㊁7岁的儿童存在该疾病[15]㊂一项中国队列研究显示,三分之一的GS患者合并有糖尿病[16]㊂此外,还有报道提到了GS患者存在软骨钙沉着症㊁肾钙沉着症㊁腘绳肌钙化等情况,表现为关节和肌肉疼痛以及蛋白尿和乏力等,可能是焦磷酸钙晶体沉积在软骨㊁腘绳肌和肾小管中所致[10]㊂4㊀GS治疗和预后㊃5391㊃GS的治疗应个体化㊂在饮食方面上,多摄入富含钾的食物如香蕉㊁土豆㊁橘子等㊂需要终身补充钾时,首选口服氯化钾㊂优先考虑补镁,因为镁的补充有助于钾的补充,并推荐剂量为4mg/kg至5mg/kg,分为4至6次㊂治疗目标为血钾ȡ3.0mmoL/l,血镁ȡ0. 6mmoL/l㊂缓释剂型更容易耐受,并需注意胃溃疡㊁腹泻等胃肠道不适情况,避免空腹服用㊂推荐根据症状㊁生物化学和副作用进行滴定式给药,分次给药㊂如病情需要,静脉补充㊂对于顽固性低钾血症患者,可考虑加用醛固酮拮抗剂㊁保钾利尿剂如螺内酯(每天200mg 至300mg)和阿米洛利(每天5mg至10mg),以及肾素-血管紧张素系统抑制剂(如缬沙坦),需警惕低血压等问题㊂患者伴有关节疼痛时推荐使用非甾体抗炎药,如吲哚美辛㊂然而基于GS的发病机制思考得知,肾素-血管紧张素系统是GS的一种保护性反应,使用螺内酯㊁缬沙坦抑制该系统的药物是否真的获益仍有待商讨㊂怀孕期间罹患GS人群,由于怀孕等生理刺激因素会掩盖GS症状,患者自我感觉不明显,临床治疗上可能需要增加电解质补充剂量,静脉给药的可能性增大[17]㊂如果已经替代了钾和镁,但仍有症状,使用氯化钠治疗可能会带来一些临床获益,因为从本质上来看,GS是由于氯化钠的丢失所致,但是否真正有效仍无确凿证据㊂关于GS长期预后方面所知甚少,需要考虑其对患者的长远影响,如慢性肾病㊁软骨钙沉着症㊁心律失常㊁继发性高血压和妊娠期治疗等㊂目前已知大多数GS预后良好,但乏力等严重影响患者日常生活质量和工作能力,而进展到终末期疾病(如严重心律失常或肾功能不全)的非常少见,在文献中仅有一例报道㊂5㊀总㊀结GS目前最常见的诊断形式是通过实验室发现㊂唯一公认的临床症状是不适感㊂基因检测在GS诊断中越来越被广泛应用,然而其费用较高㊂因此,在未来我们需要推动便宜实惠并普及化的基因检测方法以服务于有慢性低钾血症的患者群体㊂ʌ参考文献ɔ[1]㊀穆妮热㊃阿塔吾拉,郭艳英.Gitelman综合征并发症及常见合并症的研究进展[J].医学研究杂志,2023,52(05):177-179,114.[2]㊀Schlingmann KP,de Baaij JHF.The genetic spectrum ofGitelman(-like)syndromes[J].Curr Opin Nephrol Hyper-tens,2022,31(5):508-515.[3]㊀Viering D,Schlingmann KP,Hureaux M,et al.Gitelman-likesyndrome caused by pathogenic variants in mtDNA[J].AmSoc Nephrol,2022,33(2):305-325.[4]㊀Xu J,He J,Xu S,et al.Gitelman syndrome with graves'dis-ease leading to rhabdomyolysis:a case report and literaturereview[J].BMC Nephrol,2023,24(1):123. [5]㊀Vargas-Poussou R,Dahan K,Kahila D,et al.Spectrum ofmutations in Gitelman syndrome[J].Am Soc Nephrol,2011,22(4):693-703.[6]㊀黄晓利,龙易勤,郑敏.TRPM6基因突变导致原发性低镁血症继发低钙血症(附1例报告及文献复习)[J].中国临床神经科学,2018,26(2):206-211.[7]㊀Enriquez R,Adam V,Sirvent AE,et al.Gitelman syndromedue to p.A204T mutation in CLCNKB gene[J].Int UrolNephrol,2010,42(4):1099-1102.[8]㊀Mou L,Wu F.Simultaneous Homozygous Mutations inSLC12A3and CLCNKB in an inbred Chinese pedigree[J].Genes(Basel),2021,12(3):369.[9]㊀Kurstjens S,de Baaij JH,Bouras H,et al.Determinants ofhypomagnesemia in patients with type2diabetes mellitus[J].Eur Endocrinol,2017,176(1):11-19. [10]㊀Chotard E,Blanchard A,Ostertag A,et al.Calcium pyro-phosphate crystal deposition in a cohort of57patients withGitelman syndrome[J].Rheumatology(Oxford),2022,61(6):2494-2503.[11]㊀Verploegen MFA,Vargas-Poussou R,Walsh SB,et al.Par-athyroid hormone and phosphate homeostasis in patientswith bartter and Gitelman syndrome:an international cross-sectional study[J].Nephrol Dial Transplant,2022,37(12):2474-2486.[12]㊀Shen Q,Chen J,Yu M,et al.Multicentre study of the clini-cal features and gene variant spectrum of Gitelman syn-drome in Chinese children[J].Clin Genet,2021,99(4):558-564.[13]㊀Chen L,Chou CL,Knepper MA.Targeted single-cell RNA-seq identifies minority cell types of kidney distal nephron[J].Am Soc Nephrol,2021,32(4):886-896.[14]㊀陈丽萌,张抒扬,张磊等.Gitelman综合征诊疗中国专家共识(2021版)[J].罕见病研究,2022,1(01):56-67. [15]㊀Yu S,Wang C.Genetic Analysis of Gitelman Syndrome:Co-existence with Hyperthyroidism in a Two-year-old Boy[J].Endocr Metab Immune Disord Drug Targets,2021,21(8):1524-1530.[16]㊀Liu T,Wang C,Lu J,et al.Genotype/phenotype analysis in67Chinese patients with Gitelman's syndrome[J].AmNephrol,2016,44(2):159-168.[17]㊀Lim M,Gannon D.Diagnosis and outpatient management ofGitelman syndrome from the first trimester of pregnancy[J].BMJ Case Rep,2021,14(5):241756.㊃6391㊃。

33.Gitelman综合征概述Gitelman综合征(Gitelman syndrome,GS;OMIM 263800)是一种由肾脏远曲小管钠氯协同转运蛋白(NCC)功能障碍所致的常染色体隐性遗传病。
1966年由美国医生Gitelman首先报道了该病,但直至1996年其致病基因SLC12A3才得以明确。
主要临床特点为肾性失钾导致的低钾血症、代谢性碱中毒,常伴有低血镁、低尿钙和肾素-血管紧张素-醛固酮系统(RAAS)活化,血压正常或偏低。
病因和流行病学Gitelman综合征是由编码噻嗪类利尿剂敏感的钠氯协同转运体(NCC)的SLC12A3基因突变所致。
生理情况下,通道蛋白NCC位于肾脏远曲小管上皮细胞的管腔侧,参与肾小球滤过液中5%~10%氯离子和钠离子的重吸收,是机体维持水、电解质平衡的一道重要防线。
当基因突变导致NCC结构和(或)功能障碍时,氯离子和钠离子从远端肾小管重吸收减少,肾脏重吸收水减少,继发性RAAS活化、肾性失钾和钙重吸收减少。
目前在Gitelman综合征患者中已发现近500种SLC12A3基因突变(/ac/gene.php?gene= SLC12A3)。
此外,编码氯离子通道ClC-Kb的CLCNKB基因突变(Batter综合征Ⅲ型)和编码肝转录因子1-β(HNF1-β)的HNF1B基因突变也可产生类似临床表现。
Gitelman综合征是最常见的遗传性肾小管疾病之一,患病率约为1/40 000~1/4000,亚洲人群中可能更高。
由于该病易被漏诊或误诊,很难确定一般人群中该病的真实患病率,目前没有观察到男性和女性发病率的显著差异。
临床表现Gitelman综合征常于青少年或成年早期起病。
临床表现主要与低血钾和低血镁相关,轻型患者可无症状或表现为轻度乏力和纳差;严重患者会出现四肢抽搐、软瘫、痛性痉挛、晕厥和横纹肌溶解继发急性肾损伤,甚至因为严重室性心律失常导致心脏骤停。
目前认为,Gitelman综合征临床表现的异质性不仅与基因突变类型和修饰基因相关,还与患者性别和饮食习惯等环境因素相关。
Gitelman综合征诊疗中国专家共识(2021版)一、临床表现与诊断GS常见临床症状为低血钾、低血镁在全身多系统的表现,常累及骨骼肌、肾脏、胃肠道、心血管和神经系统,儿童GS患者常见就诊原因为手麻、肌无力、生长发育迟缓、手足抽搐等。
GS患者还可出现蛋白尿和肾功能损害,肾脏病理除球旁器增生外,还可表现为慢性肾小管间质损伤,少数病例合并有其他肾小球疾病。
GS合并肾小球损害的病例包括局灶节段性肾小球硬化、C1q肾病、微小病变肾病等。
二、临床诊断(1)根据患者病史排除消化道钾摄入不足或腹泻、使用利尿剂、细胞内外钾分布异常等情况。
(2)存在肾性失钾及低钾血症相关临床表现,可伴有低镁血症或低钙尿症。
肾性失钾:血钾<3.0 mmol/L时,尿钾排泄量>20 mmol/24 h; 或血钾<3.5 mmol/L时,尿钾排泄量>25 mmol/24 h。
(3)血压正常或偏低。
(4)代谢性碱中毒。
1、与Bartter综合征鉴别诊断经典的Bartter综合征(Bartter综合征Ⅲ型)由编码氯离子通道ClC-Kb的CLCNKB基因突变所致,患者发病相对较早(多在3岁前),更易出现生长发育迟缓,血镁水平多正常,尿钙水平正常或偏高。
行氯离子清除试验,若患者对氢氯噻嗪试验有反应而对呋塞米试验无反应,将有助于临床诊断Bartter综合征,基因检测可进一步鉴别。
Gitelman综合征具体诊疗流程见图1。
2. 基因诊断针对SLC12A3基因的直接测序仍是目前使用最广泛的检测方法,但约8%~30%的患者仅可检测到单杂合突变,需进一步对内含子突变及基因大片段缺失和重复进行分析。
二代测序技术、MLPA和微阵列比较基因组杂交技术(aCGH) 逐渐用于诊断GS。
对一代测序仅有SLC12A3单杂合突变的患者,建议进一步行MLPA、全外显子组或全基因组二代测序寻找其他可能的变异位点,如条件允许可直接采用二代测序技术进行基因诊断。


㊃病例报告㊃通信作者:李小青,E m a i l :x a _l x q@163.c o m S L C 12A 3基因突变G i t e l m a n 综合征并身材矮小1例临床分析许金腾1,马 凯2,李小青2,杨 颖3(1.西安医学院,陕西西安710021;2.西安市儿童医院免疫科,陕西西安710003;3.陕西省儿科疾病研究所,陕西西安710003) 摘 要:G i t e l m a n 综合征(G i t e l m a n s yn d r o m e ,G S )又被称为家族性低钾低镁血症,是一种以低钾低氯性碱中毒㊁低镁血症㊁低尿钙㊁高肾素活性为特征的常染色体隐性遗传的失盐性肾小管疾病,血压可正常或偏低㊂大多数患者经过 食补+药物 替代治疗有良好的预后,早期发现并予相应治疗,可显著提高患者生活质量㊂现将西安市儿童医院收治的1例G i t e l m a n 综合征并身材矮小患者临床资料及诊疗过程,结合相关文献学习,做如下报道㊂关键词:G i t e l m a n 综合征;低血钾;矮小症;S L C 12A 3基因中图分类号:R 696.2 文献标志码:A 文章编号:1004-583X (2019)10-0933-04d o i :10.3969/j.i s s n .1004-583X.2019.10.014C l i n i c a l a n a l y s i s o fG i t e l m a n s yn d r o m e a n d s h o r t s t a t u r ew i t hS L C 12A 3m u t a t i o n i no n e c a s e X u J i n t e n g 1,M aK a i 2,L iX i a o q i n g 2,Y a n g Y i n g31.X i a n M e d i c a lU n i v e r s i t y ,X i 'a n 710021,C h i n a ;2.D e p a r t m e n t o f I mm u n o l o g y ,X i 'a nC h i l d r e n 'sH o s p i t a l ,X i 'a n 710003,C h i n a ;3.S h a a n x iP r o v i n c i a l I n s t i t u t e o fP a e d i a t r i cD i s e a s e s ,X i 'a n 710003,C h i n aC o r r e s p o n d i n g a u t h o r :L iX i a o q i n g ,E m a i l :x a _l x q @163.c o m A B S T R A C T :G i t e l m a n s y n d r o m e (G S ),a l s ok n o w na sf a m i l i a lh y p o k a l e m i ch y p o m a gn e s e m i a ,i sa na u t o s o m a l r e c e s s i v e i n h e r i t e da n ds a l t -l o s i n g r e n a lt u b u l a rd i s e a s ec h a r a c t e r i z e db y h y p o k a l e m i ca n dh y p o c h l o r e m i ca l k a l o s i s ,h y p o m a g n e s e m i a ,h y p o c a l c i u r i a a n dh i g h r e n i n a c t i v i t y .A n dG S p a t i e n t sm a y ha v e n o r m a l o r l o wb l o o d p r e s s u r e .M o s t p a t i e n t sh a v e g o o d p r o g n o s i sa f t e r f o o ds u p p l e m e n t +d r u g r e p l ac e m e n t t h e r a p y .E a r l yde t e c t i o na n dc o r r e s p o n d i n gt r e a t m e n t c a ns i g n i f i c a n t l y i m p r o v et h el i f e q u a l i t y o f p a t i e n t s .T o g e t h e r w i t ho t h e rr e l a t e dl i t e r a t u r e ,t h i s p a p e r r e p o r t s t h e c l i n i c a l d a t a ,d i a g n o s i sa n dt r e a t m e n to f a p a t i e n tw i t hG i t e l m a ns y n d r o m ea n ds h o r t s t a t u r ea d m i t t e dt o X i 'a nC h i l d r e n 'sH o s pi t a l .K E Y W O R D S :G i t e l m a n s y n d r o m e ;h y po k a l e m i a ;s h o r t s t a t u r e ;S L C 12A 3g e n e G i t e l m a n 综合征(G i t e l m a ns y n d r o m e ,G S )又被称为家族性低钾低镁血症,是一种常染色体隐性遗传的失盐性肾小管疾病㊂主要表现为低钾低氯性碱中毒㊁低镁血症㊁低尿钙㊁高肾素活性,可有疲劳㊁夜尿增多㊁肌无力㊁肌强直,甚至发生致命性心律失常等表现,多数患儿缺乏典型临床表现,常导致漏诊㊂本文报道西安市儿童医院收治的1例G S 患者,结合相关文献复习,旨在提高临床医师对该病的认识㊂1 病例资料患儿,女,7岁10月,以 间断皮疹1月余,加重3天 于2018年10月16日收住院㊂1月余前于当地医院诊断 过敏性紫癜 住院治疗11天好转出院㊂10天前出现呕吐,伴腹痛,以下腹为主,大便呈暗红色血样便,10次/日,量中等,予 头孢哌酮钠舒巴坦钠㊁甲泼尼龙琥珀酸钠 等治疗8天症状缓解,住院期间多次查电解质:血清钾波动于2.32~2.50mm o l /L ㊂3天前再次出现典型紫癜样皮疹,伴腹痛,遂以 过敏性紫癜㊁低钾血症原因待查 转入我院㊂既往史:5岁余因动脉导管未闭行介入治疗㊂家族史:父母体健,非近亲结婚㊂查体:体温36.6ħ,呼吸20次/m i n ,血压80/55mm /H g (1mmH g =0.133k P a ),体重18.5k g ,身高100c m (<P 3)㊂意识清,精神可㊂面色欠红润㊂双眼睑无水肿㊂腹平软,无压痛及反跳痛,肝脾肋下未触及,移动性浊音阴性,肠鸣音4次/m i n ㊂脊柱㊁四肢㊁关节无红肿㊁畸形,无压痛及活动受限,双下肢可见紫癜样皮疹㊂四肢肌力㊁肌张力正常㊂余系统查体无异常㊂入院后查血常规:白细胞计数9.17ˑ109/L ,中性细胞百分比67.2%,淋巴细胞百分比21.5%,红细胞计数4.43ˑ1012/L ,血红蛋白浓度126g /L ,血小板计数550ˑ109/L ㊂尿㊁粪常规正常㊂血沉㊁降钙素㊃339㊃‘临床荟萃“ 2019年10月20日第34卷第10期 C l i n i c a l F o c u s ,O c t o b e r 20,2019,V o l 34,N o .10Copyright ©博看网. All Rights Reserved.正常㊂肝肾功能㊁甲状腺功能㊁心肌酶正常㊂凝血四项:血浆凝血酶原时间10.90s,凝血酶原活动度164.0%,凝血酶原国际标准化比值0.78,余项正常㊂血清电解质:K +2.25mm o l /L ,N a +132.0mm o l /L ,C l -94.2mm o l /L ,C a 2+2.63mm o l /L ㊂血气分析:p H7.49,p C O 227mmH g ,p O 2126mmH g ,H C O -320.60mm o l /L ,B E -2.00mm o l /L ,S p O 299%㊂血压及血糖正常㊂心电图提示:S T 段下移(V 2-V 6)㊂抗核抗体系列及抗中性粒细胞胞浆抗体:阴性㊂入院后初步诊断为 过敏性紫癜(混合型)㊁低钾血症 ㊂予口服氯化钾缓释片(0.5g /次,每天3次)㊁地塞米松抗炎及常规对症治疗后腹痛缓解㊁皮疹消退㊂期间多次复查电解质血钾均较低(表1)㊂追问病史:患儿自3岁起体格发育便落后于同龄儿㊂既往有口干㊁多饮,进食固体食物困难㊂其母亲有低血钾病史㊂完善泌尿系及双侧肾上腺超声:均正常㊂促肾上腺皮质激素10.00p g/m l (参考值10~185p g /m l ),皮质醇0.50μg /d l (参考值3.7~19.4μg/d l )㊂尿电解质4项:钾17mm o l /24h (参考值51.0~102.0mm o l /24h ),钠118.8mm o l /24h (参考值130~220mm o l /24h ),氯96.3mm o l /24h (参考值170~210mm o l /24h ),钙0.4mm o l /24h (参考值2.5~7.5mm o l /24h )㊂高血压4项:血管紧张素Ⅰ(4ħ,卧位)32.94n g /(m l ㊃h ),血管紧张素Ⅰ(37ħ,卧位)36.50n g /(m l ㊃h ),血管紧张素Ⅱ(卧位)910.40p g /m l (参考值25~60p g/m l ),肾素活性(卧位)3.56n g /(m l ㊃h )[参考值0.15~2.3n g /(m l ㊃h )],醛固酮(卧位)204p g/m l (参考值30~160p g/m l ),醛固酮/肾素活性(卧位)5.73㊂血镁正常㊂复查心电图:窦性心动过缓伴心律不齐;S T -T 改变(V 3-V 6);Q -T 间期延长542m s ㊂表1 氯化钾缓释片替代治疗后患儿血清电解质变化治疗后时间(d)血清钾(mm o l /L )血清钠(mm o l /L )血清氯(mm o l /L )血清钙(mm o l /L )口服氯化钾(3次/d )12.25132.094.22.630.5g /次32.19136.798.12.460.5g /次52.36132.693.82.500.5g /次82.70132.892.92.500.5g /次112.27132.593.52.591.0g /次152.42132.491.02.341.0g /次192.62132.790.72.461.5g /次202.75132.391.22.651.5g/次 注:参考区间:血钾3.5~5.3mm o l /L ,血钠137~147mm o l /L ,血氯99~110mm o l /L ,血钙2.25~2.67mm o l /L入院后予补钾治疗后血钾水平仍较低,且患儿无呕吐㊁腹泻,无大汗等症状,完善相关检查,逐步排除因摄入不足㊁库欣综合征㊁原发性醛固酮增多症㊁自身免疫性疾病等导致的低钾血症可能㊂考虑存在离子通道相关性疾病,征得家长同意行基因检测,结果示:S L C 12A 3基因c .911C>T (p.T 304M )和c .1129_1140d e l (p .M 377_F 380d e l )复合杂合变异(图1)㊂结合患儿临床特点及实验室检查,确诊为G i t e l m a n 综合征㊂予口服氯化钾缓释片(1.5g/次,3次/d ),复查心电图正常,血清电解质血钾较前回升,好转出院㊂出院3个月后电话随访患儿目前口服氯化钾缓释片(1.5g /次,3次/d ),血清钾维持在3.0mm o l /L ,心电图正常㊂未诉不适,病情平稳,生活如常㊂图1 受检者S L C 12A 3基因c .911C >T (p .T 304M )变异S a n ge r 测序家系验证结果,从左到右依次为患儿㊁母亲㊁父亲㊃439㊃‘临床荟萃“ 2019年10月20日第34卷第10期 C l i n i c a l F o c u s ,O c t o b e r 20,2019,V o l 34,N o .10Copyright ©博看网. All Rights Reserved.2讨论G S的病因现已明确,是位于16号染色体长臂(16q13)的溶质载体家族12成员3突变所致,其所对应的S L C12A3基因编码噻嗪类利尿剂敏感的钠氯共同转运体(N C C T)结构和/或功能异常,引起肾脏远曲小管对钠氯重吸收减少,引起低血容量,继而导致肾素-血管紧张素-醛固酮系统(R A A S)激活㊁低血钾性碱中毒等一系列病理生理和临床表现[1-2]㊂目前为止,在G S患者中已经发现有180多种噻嗪类敏感的N C C T基因(S L C12A3)突变㊂据报道18%~40%的可疑G S患者只携带一个突变基因,在对448名患者进行基因检测分析提示总突变率为88%,其中70%的患者存在2种突变,18%的患者有1种突变,12%的患者没有发现突变,这与国外学者在中国的研究结果相似[3-6]㊂国外学者对中国G S患者基因分析发现T h r60M e t为最常见突变类型[5-6],该患儿携带S L C12A3基因复合杂合变异,经S a n g e r 验证,发现c.911C>T(p.T304M)杂合变异来源于母亲,该变异为H GM D收录的已知变异: B o u c h i r e b㊁M i a o和Z h o n g等[7-9]分别发现3例G S 合并其他疾病的患者携带该杂合变异(另一变异分别为p.T60M㊁p.G439S㊁p.L700P);E x A C数据库记录其频率为0.000008;生物信息分析软件S I F T㊁P o l y P h e n-2和M u t a t i o n_T a s t e r均预测其可能有害㊂根据现有证据,该变异为疑似致病性变异㊂患儿及其父亲均携带c.1129_1140d e l(p.M377_F380d e l)变异未见文献报道;未在正常人群数据库(E x A C数据库㊁T O P M E D数据库㊁1000数据库)中检出;根据现有证据,该变异为临床意义未明的变异㊂多数G S患者于青少年或成年时期发病,其临床表现多为非特异性,轻者可无症状,或仅表现为轻度的疲劳㊁夜尿增多㊁肌无力或肌肉痉挛,重者可出现肌强直㊁麻痹㊁横纹肌溶解,甚至发生致命性心律失常㊂常见的肾外并发症为身材矮小㊁热性惊厥㊁甲状腺功能紊乱(甲状腺功能亢进或低下)㊁癫痫㊁Q-T间期延长[10-12]㊂国外学者对185例基因确诊的G S患者进行研究分析发现,54.7%的患者是由于偶然的血液检测发现,32.6%的患者因为手足抽搐得到进一步诊治,而身材矮小和具有相关家族史的患者的诊治机率分别为7.2%和5.5%[12],典型的患者可通过临床表现和实验室检查得到临床诊断,而最终确诊有赖于基因检测㊂G S尚无有效的治疗措施,目前的治疗原则为综合㊁替代治疗㊂使血钾和血镁水平分别维持在3.0 mm o l/L及0.6mm o l/L以上㊂本例患儿伴有明显的低血钾,轻度的低钠㊁低氯血症,无低镁血症,予口服氯化钾缓释片及进食适量含氯化钠的食物纠正电解质紊乱㊂同时,该患者体格发育明显落后于正常同龄儿,考虑为G S导致的生长迟缓,其确切病因尚不清楚㊂实验研究发现,低钾饮食的小鼠,身长明显缩短,体重增加㊂血清生长激素(G H)和胰岛素样生长因子1(I G F-1)水平较低[13],这提示缺钾可能影响垂体分泌G H㊂本例患儿父亲身高170c m,母亲身高168c m,患儿遗传身高为162.5c m㊂现身高100 c m(<P3),体重18.5k g(<P3),B M I值:18.5k g/ m2(P25-50),体格发育明显落后于正常同龄儿㊂据国外文献报道1例生长激素完全缺乏的G S患儿,该患者为12.9岁女童,身高138.4c m(<P3),体重43.3 k g(P25-P50),B M I22.7k g/m2(P85-P95),携带c. 494A>T(p.G l n165L e u)和c.1868T>C(p. L e u623P r o)的S L C12A3基因杂合突变,予生长激素㊁氧化镁和氯化钾替代治疗7个月,身高增长5.6 c m,临床症状缓解[14]㊂G S伴G H缺乏的患者,早期接受G H治疗生长反应良好,平均生长速度可达10.2~12c m㊂因此,对G S患儿表现生长迟缓,建议完善G H-G F I-1轴检测,必要时予生长激素替代治疗㊂此外,该患儿由于长期低钾血症导致继发性Q-T间期延长,更应加强长期随访管理,防止发生严重心律失常危及生命㊂该病是一种起病隐匿㊁进展缓慢的隐性遗传的失盐性肾小管疾病,目前仅有2例预后不良,发展为尿毒症[15-16]㊂大多数患者常因临床表现不典型,不能引起患者及医生的注意,容易导致漏诊㊂通过本病例复习,提示临床医师对于反复发作或持续存在的低钾血症患儿,尤其有家族史者,需考虑存在G S可能㊂应尽早行基因检测,早期明确诊断,早期积极干预,建立长期随访管理,可明显提高患儿生活质量,改善远期预后㊂同时,对高危产妇做好遗传咨询工作,提高家庭人口素质㊂参考文献:[1]S i m o nD,N e l s o n C,B i a M,e ta l.G i t e l m a n'sv a r i a n to fB a r t t e r's s y n d r o m e,i n h e r i t e dh y p o k a l a e m i c a l k a l o s i s,i s c a u s e db y m u t a t i o n s i n t h e t h i a z i d e-s e n s i t i v eN a-C lc o t r a n s p o r t e r[J].N a tG e n e t,1996,12(1):24-30.[2] N a k h o u lF,N a k h o u l N,D o r m a n E,e t a l.G i t e l m a n's㊃539㊃‘临床荟萃“2019年10月20日第34卷第10期 C l i n i c a l F o c u s,O c t o b e r20,2019,V o l34,N o.10Copyright©博看网. All Rights Reserved.s y n d r o m e:a p a t h o p h y s i o l o g i c a l a n d c l i n i c a l u p d a t e[J].E n d o c r i n e,2011,41(1):53-57.[3] V a r g a s-P o u s s o uR,D a h a n K,K a h i l a D,e ta l.S p e c t r u m o fm u t a t i o n s i n G i t e l m a n s y n d r o m e[J].J A m S o c N e p h r o l, 2011,22(4):693-703.[4] B a l a v o i n e A S,B a t a i l l e P,V a n h i l l e P,e t a l.P h e n o t y p e-g e n o t y p ec o r r e l a t i o n a n d f o l l o w-u p i n a d u l t p a t i e n t s w i t hh y p o k a l a e m i ao fr e n a lo r i g i ns u g g e s t i n g G i t e l m a ns y n d r o m e[J].E u r JE n d o c r i n o l,2011,165(4):665-667.[5] M aJ,R e n H,L i n L,e ta l.G e n e t i cf e a t u r e so f C h i n e s ep a t i e n t s w i t h g i t e l m a n s y n d r o m e:s i x t e e n n o v e l S L C12A3 m u t a t i o n s i d e n t i f i e d i n a n e wc o h o r t[J].A mJN e p h r o l,2016, 44(2):113-121.[6] Z e n g Y,L iP,F a n g S,e ta l.G e n e t i ca n a l y s i so fS L C12A3G e n e i nC h i n e s e P a t i e n t sw i t hG i t e l m a n s y n d r o m e[J].M e d S c iM o n i t,2019,25:5942-5952.[7] B o u c h i r e bK,B o y e r O,M a n s o u r-H e n d i l iL,e ta l.F a n c o n is y n d r o m e a n d s e v e r e p o l y u r i a:a nu n c o mm o nc l i n i co b i o l o g i c a l p r e s e n t a t i o no f aG i t e l m a n s y n d r o m e[J].B M CP e d i a t r,2014, 14:201.[8] M i a o Z,G a o Y,B i n d e l s R J M,e t a l.C o e x i s t e n c e o fn o r m o t e n s i v e p r i m a r y a l d o s t e r o n i s m i n t w o p a t i e n t s w i t hG i t e l m a n's s y n d r o m e a n d n o v e l t h i a z i d e-s e n s i t i v e N a-C lc o t r a n s p o r t e rm u t a t i o n s[J].E u r JE nd o c r i n o l,2009,161(2):275-283.[9] Z h o n g F,Y i n g H,J i aW,e t a l.C h a r a c t e r i s t i c s a n d f o l l o w-u po f13p e d i g r e e s w i t h G i t e l m a ns y n d r o m e[J].J E n d o c r i n o lI n v e s t,2018(1).[10] G i t e l m a n综合征诊治专家共识协作组.G i t e l m a n综合征诊治专家共识[J].中华内科杂志,2017,56(9):712-716. [11] B l a n c h a r dA,B o c k e n h a u e rD,B o l i g n a n oD,e ta l.G i t e l m a ns y n d r o m e:c o n s e n s u s a n d g u i d a n c e f r o m a K i d n e y D i s e a s e:I m p r o v i n g G l o b a l O u t c o m e s(K D I G O)C o n t r o v e r s i e sC o n f e r e n c e[J].K i d n e y I n t,2017,91(1):24-33.[12] U j i m u r a J,N o z uK,Y a m a m u r aT,e t a l.C l i n i c a l a n d g e n e t i cc h a r a c t e r i s t i c s i n p a t i e n t sw i t hG i t e l m a n s y nd r o m e[J].K i d ne yI n tR e p,2019,4(1):119-125.[13] G i l-P eña H,G a r c i a-L o p e z E,A l v a r e z-G a r c i a O,e t a l.A l t e r a t i o n so f g r o w t h p l a t ea n da b n o r m a l i n s u l i n-l i k e g r o w t hf a c t o r Im e t a b o l i s m i ng r o w t h-r e t a r d e dh y p o k a l e mi c r a t s:e f f e c to f g r o w t h h o r m o n e t r e a t m e n t[J].A m J P h y s i o l R e n a l P h y s i o l,2009,297(3):639-645.[14] M i n S R,C h o H S,H o n g J,e t a l.G i t e l m a n s y n d r o m ec o m b i n e dw i t hc o m p l e t e g r o w t hh o r m o n edef i c i e n c y[J].A n nP e d i a t rE n d o c r i n o lM e t a b,2013,18(1):36-39.[15] B o n f a n t e L,D a v i s P A,S p i n e l l o M,e t a l.C h r o n i c r e n a lf a i l u r e,e n d-s t ag e r e n a l d i s e a s e,a n d p e r i t o n e a l d i a l y s i si nG i t e l m a n's s y n d r o m e[J].A mJK i d n e y D i s,2001,38(1):165-168.[16] C a lòL A,M a r c h i n iF,D a v i sP A,e ta l.K i d n e y t r a n s p l a n t i nG i t e l m a n's s y n d r o m e.R e p o r t o f t h e f i r s t c a s e[J].JN e p h r o l,2003,16(1):144-147.收稿日期:2019-07-08编辑:张卫国㊃639㊃‘临床荟萃“2019年10月20日第34卷第10期 C l i n i c a l F o c u s,O c t o b e r20,2019,V o l34,N o.10Copyright©博看网. All Rights Reserved.。

gitelman综合征诊断标准
Gitelman综合征是一种遗传性肾小管功能障碍疾病,其诊断主要依靠以下几个标准:
1. 临床表现:Gitelman综合征的主要特征是低血镁、低血钾、代谢性碱中毒和多尿。
患者可能表现为肌肉痉挛、脱力、心动过缓、心律失常和易激惹等症状。
2. 实验室检查:血液和尿液测试可以发现低血镁、低血钾、碱中毒、尿钾排泄过多等异常结果。
这些异常结果可以帮助确定Gitelman综合征的诊断。
3. 基因检测:根据临床症状和实验室结果,可以进一步进行基因检测,通过检测SLC12A3基因的突变来确认Gitelman综合征的诊断。
需要强调的是,由于Gitelman综合征的临床表现和实验室结果可能与其他肾小管功能障碍疾病相似,且该疾病的表现和严重程度会有所不同,因此确诊Gitelman综合征需要综合考虑临床症状、实验室结果以及基因检测等多个因素。
诊断和治疗应由专业医生进行。

Bartter综合征与Gitelman综合征的诊断与治疗Bartter综合征和Gitelman综合征都是罕见的遗传性疾病,它们都会导致肾脏失去过多的电解质,特别是钾离子。
这会导致一系列的症状,如肌肉无力、疲劳、头晕等。
对于Bartter综合征,诊断通常是通过基因检测来确定的。
我们会检测患者的基因是否出现了突变。
如果有突变,那么就可以确诊为Bartter综合征。
我们还会进行血液和尿液的电解质检查,以确定患者的电解质水平是否偏低。
在治疗方面,Bartter综合征目前没有根治的方法,但是我们可以通过药物来控制症状。
例如,我们会使用保钾利尿剂来减少钾的流失,使用血管紧张素转换酶抑制剂来减少醛固酮的分泌,从而减少钾的流失。
我们还会根据患者的症状来使用一些支持性的药物,如补充维生素D和钙剂来预防骨质疏松。
对于Gitelman综合征,诊断也是通过基因检测来确定的。
我们会检测患者的基因是否出现了突变。
如果有突变,那么就可以确诊为Gitelman综合征。
我们还会进行血液和尿液的电解质检查,以确定患者的电解质水平是否偏低。
在治疗方面,Gitelman综合征的治疗方法和Bartter综合征类似。
我们也会使用保钾利尿剂来减少钾的流失,使用血管紧张素转换酶抑制剂来减少醛固酮的分泌,从而减少钾的流失。
我们还会根据患者的症状来使用一些支持性的药物,如补充维生素D和钙剂来预防骨质疏松。
总的来说,Bartter综合征和Gitelman综合征虽然是一种罕见的疾病,但是通过基因检测和电解质检查,我们可以准确地诊断出这种疾病。
而且,通过使用保钾利尿剂、血管紧张素转换酶抑制剂和支持性的药物,我们可以很好地控制患者的症状,提高患者的生活质量。
重点和难点解析:在处理Bartter综合征和Gitelman综合征的诊断与治疗过程中,有几个关键的细节需要我们特别关注。
这些细节对于确保准确诊断和有效治疗至关重要。
基因检测在诊断中的作用不容忽视。
Bartter综合征和Gitelman 综合征都是遗传性疾病,因此,通过基因检测可以确定患者是否携带有突变基因,从而确诊疾病。

从1例Gitelman综合征探讨低钾血症的全科诊疗思路
朱凯瑞;李霞;李晓岚
【期刊名称】《巴楚医学》
【年(卷),期】2022(5)2
【摘要】血清钾浓度<3.5 mmol/L称为低钾血症(hypokalemia)。
多数低钾血症患者无症状或症状不典型。
当患者反复出现低钾血症或合并其他疾病,口服补钾效果欠佳时应住院治疗。
Gitelman综合征(Gitelman syndrome,GS)是罕见的家族性低钾-低镁血症,随着诊断技术的进步,临床病例报道越来越多,GS的患病率为(1~10)/4万,亚洲人群患病率较高[1]。
【总页数】4页(P14-17)
【作者】朱凯瑞;李霞;李晓岚
【作者单位】新疆医科大学第五附属医院全科医学科;新疆医科大学第五附属医院内分泌科
【正文语种】中文
【中图分类】R442.8
【相关文献】
1.基于门诊就诊疾病谱分析的综合医院全科医疗科在分级诊疗中的定位与发展探讨
2.基于全科团队和分级诊疗的基层健康管理模式优化探讨
3.老年骨质疏松症的全科诊疗思路
4.基于“生物-心理-社会”模式的甲状腺癌术后患者一例报道及全科诊疗思路
5.消瘦的全科诊疗思路
因版权原因,仅展示原文概要,查看原文内容请购买。

中国乡村医药近亲婚配生育的Gitelman综合征1例杨静叶玉燕陈丽萍朱志伟方霞王朝晖Gitelman综合征是家族性低血钾、低血镁、低尿钙症,合并低氯性碱中毒,伴有正常或偏低的血压,并有可能激活肾素-血管紧张素-醛固酮系统,是较常见的一类遗传性原发性肾小管疾病。
患者临床多表现为乏力、四肢麻木或抽搐、肌肉痉挛等,部分患者伴有夜尿增多,严重者并发软骨钙质沉积、肾功能不全、室性心律失常甚至心源性猝死。
Gitelman综合征一般由编码肾远曲小管噻嗪类利尿剂敏感的钠氯共同转运体(NCCT)的SLC12A3基因发生突变引起[1]。
目前,基因突变分析仍为诊断Gitelman综合征的金标准。
现回顾性分析近亲婚配(表兄妹)生育的Gitelman综合征1例的临床资料及基因检测结果,探讨SLCl2A3基因突变类型及遗传特点,旨在提高临床医师对Gitelman综合征的认识,避免误诊和漏诊。
1 病历摘要患者女,25岁,因“乏力不适”于2017年3月1日至当地医院就诊,血钾低至2.65mmol/L,予以补钾治疗半年后仍反复低钾,遂于9月25日转至金华市人民医院肾内科进一步诊治。
患者身高152cm,体重52kg,血压正常,生长发育正常,精神可,甲状腺未扪及肿大,心肺腹部未见明显异常,四肢无水肿。
追问病史,患者近2年来每晚夜尿3~4次,间断性腹泻,有口干、眼干不适,时有耳鸣。
患者既往体健,否认长期服用利尿剂、缓泻剂史,否认毒物暴露史、家族性遗传病史。
患者无兄弟姐妹,其父母为表兄妹近亲婚配。
父母化验血钾均于正常范围。
彩超示肝、胆、胰、脾、双肾、双侧肾上腺区未见明显异常;膀胱未见明显异常;双侧输尿管未见明显扩张。
血常规、乙肝三系、肝肾功能、血糖、皮质醇节律、甲状腺功能、抗核抗体、抗可提取性核抗原抗体、抗双链DNA抗体、免疫球蛋白、立卧位肾素活性、血管紧张素Ⅱ、醛固酮等未见异常。
血电解质:钾3.02mmol/L,钠135mmol/L,氯95mmol/L,镁0.73mmol/L,钙,磷正常。
Gitelman综合征:附1例报道并文献复习
陈芬琴;王涤非;刘珂;吴坤艳;张艳玲;赵岩
【期刊名称】《中国医科大学学报》
【年(卷),期】2016(045)006
【摘要】Gitelman综合征是一种少见性遗传性疾病,本文通过回顾性分析1例Gitelman综合征患者临床表型特征及基因测序,发现Gitelman综合征可能存在SLC12A3基因外的突变,需要更多的基因筛查研究加以证实.
【总页数】4页(P563-566)
【作者】陈芬琴;王涤非;刘珂;吴坤艳;张艳玲;赵岩
【作者单位】中国医科大学附属第一医院老年病科,沈阳110001;中国医科大学附属第一医院老年病科,沈阳110001;中国医科大学附属第一医院老年病科,沈阳110001;中国医科大学附属第一医院老年病科,沈阳110001;中国医科大学附属第一医院老年病科,沈阳110001;中国医科大学附属第一医院老年病科,沈阳110001【正文语种】中文
【中图分类】R596.1
【相关文献】
1.儿童Gitelman综合征1例报道并文献复习 [J], 王小军;杜东海;石凯丽
2.Gitelman综合征1例报道并文献复习 [J], 高海波;苏晓清;张雅薇;龚玉萍;谢勇丽;赖轻舟;袁鹏;朱明慧;吴小琴;潘幸
3.Gitelman综合征1例报道并文献复习 [J], 何俊俊;陈月平;赵咏莉;华强;孟祥健;李勤;张斌华;高家林
4.SLC12A3基因突变致Gitelman综合征一家系报道并文献复习 [J], 朱智峰;闫朝丽
5.Gitelman综合征合并二次妊娠1例报道并文献复习 [J], 陈国庆;乔宠
因版权原因,仅展示原文概要,查看原文内容请购买。
Gitelman综合征诊疗中国专家共识(2021版)【摘要】近年来,国内对Gitelman综合征的研究取得了长足进步,中国人群Gitelman综合征新的临床表型、疾病分型、改良功能试验、生物标志物和特殊人群管理经验相继在国内外期刊报道。
基于最新循证医学证据,多学科领域专家在Gitelman综合征的临床表型、诊断、治疗、慢性并发症及合并症管理方面达成一致共识,为临床规范化诊疗提供了重要依据。
1共识制订方法2020年7月20日,“Gitelman综合征诊疗专家共识研讨会”召开,多学科专家讨论了共识的总体框架,并进行具体任务分工;基于标准的疾病模式(临床表现、诊断、疾病分级与分型、治疗),并结合GS研究最新进展,确定了共识拟解决的问题;组建了共识制订工作组,包括专家组、秘书组和编写组,其中专家组成员来自肾内科、心内科、儿科、内分泌科、产科、麻醉科、检验科、遗传学和临床流行病学等领域。
共识编写组以“Gitelmansyndrome”“Gitelman综合征”“吉特曼综合征”为主题词检索了PubMed、Embase、CochraneLibrary、ClinicalTrials、中国知网、万方数据知识服务平台和中文维普数据库,检索时间为建库至2020年7月10日,经去重处理后共获得相关文献1634篇,阅读文献摘要和/或全文,最终纳入符合本共识主题的文献76篇。
依据牛津循证医学中心证据分级方法并参考国内外共识制订的方法学文献[3-4],将纳入的研究证据分为6个证据等级(1a、1b、2a、2b、3、4)(表1),并根据证据等级将推荐强度分为A(强)、B(中)、C(弱)3级(表2),以代表共识制订专家组的建议。
2021年5月初,编写组完成了共识意见初稿;5月15日,于北京举行了共识专家组会议;7月5日,专家组进行线上交流、沟通,逐条讨论、修改和完善共识意见;7月23日,所有条款均经专家组无记名投票,赞成人数在85%以上被认为专家意见达成共识[5],最终形成共识终稿。
Gitelman综合征一例报告作者:刘崇斌潘银凤孙洪涛来源:《饮食与健康·下旬刊》2016年第07期【摘要】本文将对Gitelman综合征病例做一报告。
【关键词】Gitelman 综合征;电解质紊乱;低血钾;低血镁1.临床资料患者,男,24岁,教师,主因“间断心悸伴四肢抽搐1年,加重5小时”入院。
现病史:患者于1年前无明显诱因出现心悸伴四肢抽搐、乏力,经休息20分钟后症状不缓解,就诊当地医院。
相关检验:血清钾2.4mmol/L、血清氯80mmol/L,并给予口服及静脉补钾,治疗后上述症状明显缓解。
此后规律氯化钾缓释片(3.0g/次 3次/日)、螺内酯片(40mg 2次/日),并定期复查血钾离子,多维持在3.0mmol/L左右,病因诊断不清。
患者于5小时前无明显诱因再次出现上述症状,经休息症状不缓解,急来我院门诊,查离子示血清钾1.82mmol/L,门诊诊断“电解质紊乱-低钾血症”,给予口服“10%KCl注射液30ml”后症状缓解,门诊以“低钾血症”收入我科室。
家族史:其兄长39岁时猝死,死因不详。
入院查体:体温37.5摄氏度,呼吸20次/分,脉搏99次/分,血压129/78mmHg。
神清语利,步入病房,查体合作。
双肺无罗音,心界不大,心率99次/分,心律齐,心脏各瓣膜听诊区未闻及病理性杂音。
腹平软,无压痛及反跳痛,肝脾肋下未触及,肠鸣音4次/分。
双下肢无水肿,病理反射未引出。
辅助检查:离子示血清钾为2.47mmol/L,血清钠128mmol/L,血清氯103.80mmol/L。
钙2.38mmol/L,镁0.47mmol/L,磷0.11mmol/L。
尿电解质检查24小时示尿钙明显降低,24小时尿钙1.15mmol/L,尿钾为66mmol/L。
尿素6.20mmol/L,血肌酐84umol/;肝功、血流变动、心肌标志物、血常规未见异常;RAAS升高,其血浆中立位肾素活性(PRA)5.02(mL· h)、卧位肾素活性为3.0(mL· h),,立位血管紧张素II 588.6 pg/ mL、卧位血管紧张素II 27.96 pg/ mL,立位血浆醛固酮386.4 pg/ mL、卧位血浆醛固酮 146.17 pg/ mL;甲状腺功能T3、T4未见异常;甲状腺彩超未见异常;肾上腺64排螺旋CT未见明显异常;行肾脏穿刺活检:肾小球旁器细胞中度增生。
Gitelman综合征SLC12A3基因突变分析及分子机制研究王春莉;郑必霞;周玮;车若琛;赵非;张爱华;丁桂霞【期刊名称】《罕见病研究》【年(卷),期】2024(3)1【摘要】目的回顾性分析2015年8月至2022年11月南京医科大学附属儿童医院收治的20例Gitelman综合征患儿临床症状及基因突变情况,初步探讨中国人群高频突变D486N致病分子机制。
方法收集患儿的临床资料及SLC12A3基因变异情况等,在人胚胎肾293T细胞(HEK293T)中分别过表达野生型和变异型SLC12A3基因,使用蛋白免疫印迹法和免疫荧光技术分别检测肾噻嗪敏感性钠-氯协同转运体(NCC)的表达水平和亚细胞定位,探讨SLC12A3基因高频突变D486N对NCC蛋白表达和定位的影响。
结果本研究期间共收集到20例Gitelman综合征患者,患儿均表现为低血钾症,共筛查到26种SLC12A3基因突变,错义变异13种、同义变异1种、无义变异1种、移码变异4种、剪接位点变异7种。
其中4种突变p.T235K、c.1096-1G>A、p.A464A、c.2660+1_2660+2insT为新发突变。
结论本研究初步发现中国人群高频突变D486N影响NCC总蛋白和膜蛋白的表达,并影响NCC蛋白的膜表达。
本研究的实验结果可为Gitelman综合征遗传咨询及诊治提供实验依据。
【总页数】7页(P50-56)【作者】王春莉;郑必霞;周玮;车若琛;赵非;张爱华;丁桂霞【作者单位】南京医科大学附属儿童医院重点实验室;南京医科大学附属儿童医院肾脏科【正文语种】中文【中图分类】R692【相关文献】1.SLC12A3基因突变Gitelman综合征并身材矮小1例临床分析2.SLC12A3基因突变致Gitelman综合征一家系报道并文献复习3.1例Gitelman综合征合并亚临床甲状腺功能减退症患者SLC12A3基因突变位点鉴定及遗传分析4.SLC12A3基因突变致成人Gitelman综合征1例因版权原因,仅展示原文概要,查看原文内容请购买。
Gitelman综合征
Gitelman综合征(GS)是一种常染色体隐性遗传的失盐性肾小管疾病,临床特征为低钾代谢性碱中毒伴低镁血症和低尿钙症。
临床表现
多数GS患者于青少年或成年发病,但一些临床症状也可在儿童期甚至新生儿期出现,约1/3的患者可有明确的家族史。
GS常见的临床症状多为非特异性,常与电解质紊乱及RAAS激活等有关,包括以下表现:
全身症状
肢体乏力、疲劳、运动耐量下降、口渴、多饮、嗜盐;
心血管系统
血压正常或偏低、心悸、QT间期延长、室性心律失常;
消化系统
发作性腹痛、便秘、呕吐;
泌尿系统
多尿、夜尿、遗尿、蛋白尿、低钾性肾病;
神经-肌肉系统
头晕、眩晕、共济失调、假性脑瘤、肢体麻木、感觉异常、肌肉痉挛、抽搐、横纹肌溶解;
骨关节系统
关节痛、软骨钙质沉着症;
生长发育
发育停滞、生长迟缓、青春期延迟。
需要指出,多数GS患者尿蛋白定量正常或轻度升高,一般为中小分子蛋白,可能与长期低钾所致的肾小管损伤有关,大多数患者肾功能正常,因此无需肾穿刺活检。
但患者如果出现大量蛋白尿、原因不明的肾功能受损等,需行肾穿刺活检明确是否合并肾小球病变或其他肾
脏疾病。
实验室检查
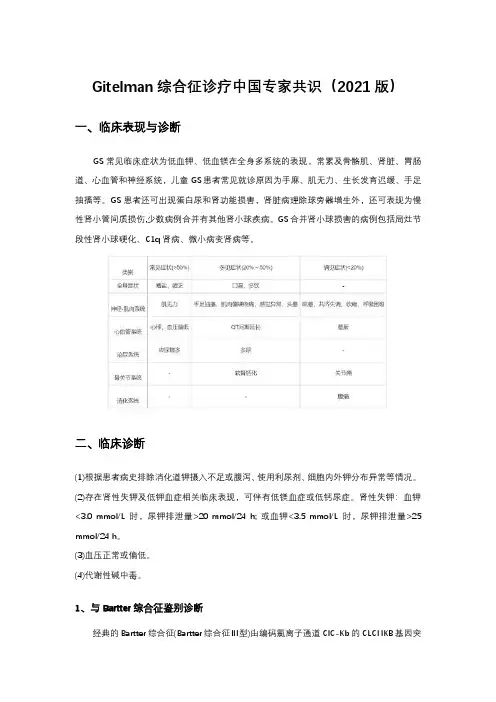
生化及影像学检查
由于GS患者症状缺乏特异性,临床诊断更多依赖于实验室检查,典型患者临床表现为"五低一高"和代谢性碱中毒,即低血钾、低血镁、低血氯、低尿钙、偏低血压和RAAS活性增高。
特别是低血镁和低尿钙对诊断GS有重要价值。
支持GS诊断的实验室检查结果主要包括:
◆低钾血症及肾性失钾:血清钾<3.5 mmol/L(严重者<2.0 mmol/L,排除使用降钾类药物),常持续存在或反复出现,伴肾性失钾(尿钾/尿肌酐>2.0 mmol/mmol或血钾低于3.5 mmol/L时24 h尿钾>25 mmol);
◆代谢性碱中毒;
◆低镁血症及肾脏排泄镁增多:血镁<0.7 mmol/L,镁排泄分数(FEMg)>4%{FEMg=[尿镁(mmol/L)×血肌酐(mmol/L)]/[血镁(mmol/L)×尿肌酐(mmol/L)]};
◆低尿钙:成人随机尿中尿钙/尿肌酐<0.2 mmol/mmol;
◆ RAAS系统激活(血浆肾素、血管紧张素及醛固酮水平增高或活性增强);
◆氯离子排泄分数(FECl)>0.5% {FECl=[尿氯(mmol/L)×血肌酐(mmol/L)]/[血氯(mmol/L)×尿肌酐(mmol/L)]} ;
◆肾脏超声检查正常(一般无钙质沉着或发育异常)。
上述检查中,血液和尿液标本需同步留取,建议留取2~3次。
如果患者正在补钾补镁治疗且血电解质水平正常或接近正常,则可停用相关药物48 h后进行检测,以免干扰检查结果。
当患者存在肾脏畸形或发育异常性疾病、出生前羊水过多、发病早于3岁、长期应用利尿剂或缓泻剂、长期高血压病史及无低钾血症或细胞外液体增加等临床表现时,则不支持GS的诊断。
氯离子清除试验(氢氯噻嗪试验)
由于GS患者的病变部位在远曲小管(氢氯噻嗪作用部位),故氯离子清除试验中,速尿能使GS患者的氯离子排泄明显增加,而氢氯噻嗪则对患者的氯离子清除影响不大,从而可以鉴别GS与Bartter综合
征(病变部位在髓襻升支粗段,为速尿作用部位)。
但氯离子清除试验过程较为复杂,且存在加重低血钾的风险。
随着基因检测技术的不断成熟,已不推荐该试验作为常规检查。
基因检测
所有患者均应行家系调查,并推荐在有条件的机构行基因检测以获得确诊。
目前世界范围内已有超过400个不同的SLC12A3基因致病突变被报道,不同地区的常见突变位点有所不同,如欧洲人中IVS9+1G>T 剪切突变多见,而我国患者中以T60M和D486N位点突变较为多见,可供基因筛查时参考。
因此,典型GS患者可通过临床表现和实验室检查获得临床诊断,而最终确诊则有赖于基因检测。
其详细的诊断标准可参考改善全球肾脏病预后组织(KDIGO)推荐的建议,见下表。