mega的使用
- 格式:pdf
- 大小:714.33 KB
- 文档页数:21

mega用法
"Mega" 的用法取決於上下文,可以是形容詞、名詞或副詞。
1. 形容詞:用來描述非常大的事物或程度很高的事物,相當於"very" 或"extremely"。
- The concert was a mega success.(這場音樂會非常成功。
)
- She has a mega talent for singing.(她有很高的唱歌天賦。
)
2. 名詞:指稱巨大的事物或大型的組織或公司。
- The company is a mega in the tech industry.(該公司是科技行業的巨頭。
)- The new shopping mall is a mega with multiple floors and hundreds of stores.(這座新購物中心有多層樓和數百家商店。
)
3. 副詞:表示以巨大或極大的程度進行某事,通常與動詞或形容詞搭配使用。
- The car sped past us mega fast.(那輛車飛速超過了我們。
)
- She is mega talented and can play multiple instruments.(她非常有才華,可以演奏多種樂器。
)。
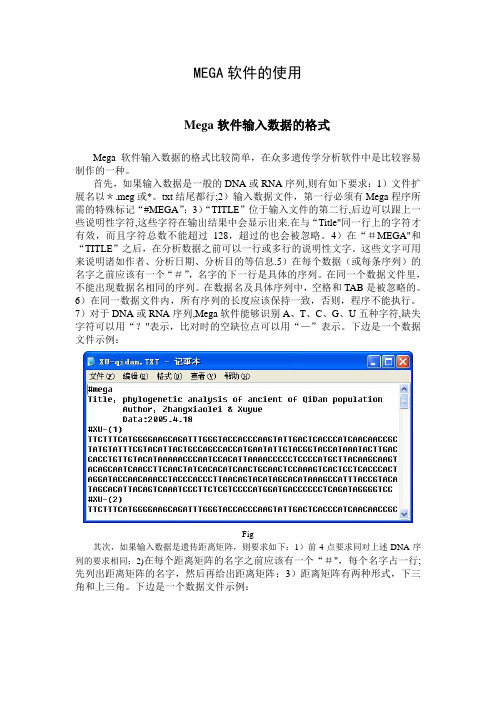
MEGA软件的使用Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*。
txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来.在与“Title"同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA"和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息.5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?"表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#",每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:Fig下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
Fig再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等.而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
MEGA的主界面:使用步骤1. 序列导入可以通过Data导入数据也可以通过file导入数据。
如果翻开的文件是比对结果,选择Analyze;如果翻开的文件是序列文件,选择Align。
另外双击这些后缀名文件即可自动导入序列,导入后会弹出MEGA比对界面。
如果fasta 序列导入报错,多是因为序列长度不同导致:如果序列长度不同,可以采用新建文件,将序列文件导入的方法。
步骤:Align →Edit/Build Alignment →create a new alignment →Data →open →Retrieve sequences from File将复制输入的序列另存输出看看。
步骤:data →Export alignment →fasta format序列长度都被用横线补齐了。
2. 多序列比对选择muscle或者clustalw进展比对:clustalw 一般用于DNA ,muscle多用于蛋白。
在比对之前需先选中要进展比对的序列〔Shift〕,还可以对序列或者序列名进展编辑〔双击〕。
比对参数选择:保存比对文件,进化树分析提供数据。
一般导出的比对结果保存为fasta格式,或者直接点击保存按钮将结果,保存为二进制的mas 或meg文件。
3. 构建进化树导入数据:将刚刚另存的meg 文件重新导入到mega程序中〔直接拖入工作界面〕,并选择构建进化树。
参数选择:参数设置,Bootstrap method一般选择1000~1500;第一次绘图时建议选择500,这样运行速度会比价快,结果适宜再调至1000重新进展进化分析。
描述:进化树可视化:View→Tree/Branch style选择树的模式,也可以通过右图菜单进展选择。
发散树环状树进化树简单美化:可视化文件的保存:•Newick——标准树文件,用于下游可视化软件的导入•png——压缩图像文件•pdf——矢量图像文件保存为png/pdf,显示不全怎么办?——将图和图注复制到WORD中保存即可〔Image Copy to Clipboard 粘贴到WORD〕。

MEGA使用说明书1. 简介1.1 MEGA的概述1.2 相关术语解释2. 安装与注册2.1 安装包并进行安装2.2 注册新账号或登录现有账号3. 用户界面导览3.. 主要功能区域介绍- 文件管理器:浏览、和文件。
- 联系人列表:查看已添加联系人信息。
- 消息中心:接收系统通知和消息更新。
4.文件操作指南4.. 浏览及搜索文件a) 使用目录树结构快速定位所需文件夹。
b) 利用搜索框输入关键词,筛选出相关文档。
c)利用标签对重要文档进行分类整理。
5.分享与协作a). 分享i)一个公开可访问的,并发送给他人以便共享特定内容。
ii)设置密码保护来限制只能被授权用户打开该。
6.数据同步a). 同步设备间数据i)在不同设备上同时登陆自己的帐户, 可实时获取最新版本资料;ii ) 手动选择需要离线保存到本地端硬盘空间内之个别项目;7.版本历史记录a). 查看文件的修改历史i)查看特定文档在不同时间点上所做过的更改;ii ) 还原到之前某个具体时刻保存下来的旧版;8. 设置与选项8.1 账户设置- 修改密码、邮箱等账户信息。
8.2 高级选项- 自定义和速度限制,以满足网络环境需求。
9.常见问题解答a) 如何恢复误删除或丢失数据?b) 如何增加存储空间容量?c) MEGA是否支持多人同时编辑一个文件?10.附件- 用户手册.pdf11.法律名词及注释i)用户:指使用MEGA服务并注册帐号进行操作和管理资料者。
ii)分享:通过公开可访问URL地址将内容共享给他人。
iii)授权用户: 指被允许浏览、编辑或已经分享出去资源的其他Mega 帐号所有者。

mega的原理及应用一、mega的定义和背景•mega是一种云存储服务,提供大容量的云端存储和共享功能。
•mega的背景:随着数字化时代的到来,个人和企业对云存储的需求越来越大,尤其是需要存储和共享大容量文件时。
二、mega的原理Mega的原理包括以下几个方面:1.分布式存储–mega采用分布式存储技术,将文件的各个部分分散存储在不同的服务器上。
–这种分布式的存储方式保证了数据的可靠性和高可用性。
2.数据加密–mega使用端到端加密来保护用户的数据安全。
–这意味着在数据传输和存储的过程中,用户的数据都是经过加密的,只有用户才能解密。
3.快速同步和共享–mega提供了快速的文件同步和共享功能,这意味着用户可以方便地将文件上传到云端,并与他人共享。
–mega通过实时同步功能,确保用户在不同设备上访问最新版本的文件。
4.强大的搜索和管理功能–mega提供了强大的搜索和管理功能,让用户可以轻松地查找和管理存储在云端的文件。
–用户可以使用关键字搜索文件,并根据不同的属性(如文件类型、修改日期等)进行排序和筛选。
三、mega的应用场景mega的应用非常广泛,主要包括以下几个方面:1.个人文件存储和备份–mega可以作为个人用户的文件存储和备份解决方案。
用户可以将自己的照片、视频、音乐等文件上传到mega,以便在不同设备间进行访问和共享。
–mega的自动同步功能还可以保证文件的及时备份,避免数据丢失的风险。
2.团队协作和共享–mega也广泛应用于团队协作和共享领域。
团队成员可以将工作文件和文档上传到mega的共享文件夹中,实现实时协同编辑和共享。
–mega的版本控制和权限管理功能,可以确保团队成员对文件的操作有序和安全。
3.学习和教育资源共享–学生和教育工作者也可以利用mega来共享学习和教育资源。
教师可以将课件、教案和学习材料上传到mega,并与学生共享。
–学生可以方便地在不同设备上访问和下载这些资源,提高学习效果。

mega使用手册Mega使用手册欢迎阅读Mega使用手册。
本文档将详细介绍如何使用Mega以及各项功能的使用方法。
请按照以下章节进行阅读。
1.简介1.1 Mega的概述1.2 Mega的优势1.3 Mega的功能2.账户管理2.1 注册Mega账户2.2 登录和注销账户2.3 密码重置和修改2.4 安全性设置3.文件和3.1 文件3.2 文件3.3 文件夹管理和操作3.4 文件共享和协作4.数据同步与备份4.1 同步文件和文件夹4.2 数据备份和恢复4.3 版本控制和历史记录5.文件搜索和过滤5.1 文件搜索功能5.2 文件过滤和筛选6.安全性和隐私6.1 文件加密和解密6.2 权限管理和共享控制6.3 数据安全和隐私保护7.终端与云端互通7.1 Mega客户端的安装和配置 7.2 云端与本地文件同步7.3 终端与云端文件交互8.其他功能8.1 文件预览和编辑8.2 多设备同步8.3 文件批量处理8.4 容量管理和空间优化附件:本文档所涉及的附件可于Mega官方网站的附件页面。
法律名词及注释:1.Mega:一种云存储和文件同步服务,由Mega Limited提供。
2.账户:用户在Mega上注册的个人或企业账户。
3.文件:用户在Mega上和存储的各种类型的文件。
4.文件夹:用户可以在Mega上创建和管理的文件夹,用于组织和管理文件。
5.文件共享:用户可以通过Mega共享,方便他人访问和文件。
6.数据同步:将本地文件同步到云端,并保持文件的最新状态。
7.数据备份:将云端数据备份到其他存储介质,以避免数据丢失。
8.文件加密:使用加密算法对文件进行加密,以保护文件的安全性和隐私性。
感谢阅读本文档,如有任何问题或困惑,请随时联系我们的客户支持团队。
MEGA软件的使用Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,打开Mega程序,有如下图所示的操作界面:单击工具栏中的“File”按钮,会出现如下图所示的菜单:从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA 格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。
单击“Open Data”选项,会弹出如下菜单:浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面:此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。
根据输入数据的类型,选择一种,点击“OK”即可。
如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示:根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。
点击“OK”按钮,即可导入数据。
如果是核苷酸数据,则读完之后,会弹出如下对话框:如上图,如果是编码蛋白质的核苷酸序列,则选择“Yes”按钮;如果是不编码蛋白质的核苷酸序列,则点击“No”按钮。
之后,会弹出如下操作窗口:此作界面的名称是“Sequence Data Explorer”,在其最上方是工具栏“Data”、“Display”、“Highlight”等,然后是一些数据处理方式的快捷按钮,在操作界面的左下方是每个序列的名称。
MEGA软件的使用Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。
在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息。
5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
1.MEGA构建系统进化树的步骤2.CLUSTALX进行序列比对1.MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
如图:2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图:。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图:。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes,如图:。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,如图:6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。
MEGA使用说明书MEGA使用说明书1、简介MEGA是一款跨平台的云存储和文件共享服务提供商。
它为用户提供了安全、稳定且易于使用的云端存储解决方案。
本文档将详细介绍MEGA的功能和使用方法,以帮助用户快速上手并充分利用该平台。
2、注册和登录2.1 注册账号2.1.1 打开MEGA官方网站2.1.2 注册按钮2.1.3 输入个人信息并创建账号2.2 登录账号2.2.1 打开MEGA官方网站2.2.2 输入注册时所使用的账号和密码2.2.3 登录按钮3、文件和3.1 文件3.1.1 在MEGA界面中找到需要的文件夹3.1.2 按钮3.1.3 选择本地文件并确认3.2 文件3.2.1 在MEGA界面中找到需要的文件3.2.2 右键文件,并选择选项3.2.3 选择本地保存路径并开始4、文件夹管理4.1 创建文件夹4.1.1 在MEGA界面中找到所需文件夹的位置4.1.2 右键文件夹的上级目录,并选择新建文件夹选项 4.1.3 输入文件夹名称并确认创建4.2 移动文件夹4.2.1 在MEGA界面中找到要移动的文件夹4.2.2 右键文件夹,并选择移动选项4.2.3 选择目标位置并确认移动4.3 删除文件夹4.3.1 在MEGA界面中找到要删除的文件夹 4.3.2 右键文件夹,并选择删除选项4.3.3 确认删除操作5、文件共享和权限管理5.1 共享文件5.1.1 在MEGA界面中找到要共享的文件 5.1.2 右键文件,并选择共享选项5.1.3 输入共享和访问权限,并共享5.2 管理共享5.2.1 在MEGA界面中找到需要管理的共享 5.2.2 右键共享,并选择管理选项5.2.3 根据需要修改共享权限或取消共享5.3 接受/拒绝共享5.3.1 在接收到共享的通知邮件中5.3.2 登录MEGA账号5.3.3 根据需求接受或拒绝共享请求6、安全和隐私设置6.1 修改密码6.1.1 登录MEGA账号6.1.2 进入个人设置界面6.1.3 找到密码修改选项并进行修改操作6.2 启用二次验证6.2.1 登录MEGA账号6.2.2 进入个人设置界面6.2.3 找到二次验证选项并进行配置6.3 隐私设置6.3.1 登录MEGA账号6.3.2 进入个人设置界面6.3.3 根据需求修改隐私设置7、恢复和备份7.1 回收站7.1.1 在MEGA界面中找到回收站选项7.1.2 找到需要恢复的文件或文件夹7.1.3 右键文件或文件夹并选择还原选项7.2 历史版本7.2.1 在MEGA界面中找到历史版本选项7.2.2 找到需要恢复的文件7.2.3 右键文件并选择历史版本选项7.2.4 选择要恢复的版本并确认恢复8、常见问题解答9、附件本文档涉及附件:- MEGA用户手册:pdf- MEGA常见问题与解答:docx10、法律名词及注释- MEGA:MEGA是MEGA Limited公司的注册商标,是一家提供云存储和文件共享服务的公司。
MEGA的使用产生背景及简介随着不同物种基因组测序的快速发展,产生了大量的DNA序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。
MEGA就是基于这种需求开发的。
MEGA 软件的目的就是提供一个以进化的角度从DNA和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。
现在我们使用的是MEGA4的版本。
它主要集中于进化分析获得的综合的序列信息。
使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。
此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。
此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。
在计算矩阵方面有一些自己的特点:1.推测序列或者物种间的进化距离2.根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树3.考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。
4.随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容可以被保存,复制。
具体使用我们以分析20个物种的血红蛋白为例来具体说明此软件的具体使用情况。
一.启动程序1.运行环境:在Windows 95/98, NT, ME, 2000, XP, vista等操作系统下均可使用。
2.下载安装:可以直接登陆进行下载安装,另外还可以从/tools/phylogeny.php中的链接进去。
3.双击桌面快捷方式图标,进入主界面;或者从开始菜单,单击图标启动。
二.序列分析。
1.启动单击后,会出现如下界面:这里有三个选项,分别对应三种不同的情况:以下分别予以介绍:Create a new alignment :是在你没有任何比对的时候使用,比如你只有一个fasta格式的序列就可以选择这个选项。
Open a saved alignment session:使用它可以打开一个我们已经比对好的序列文件;Retieve a sequence from a file :这种情况同第一种情况相似,只是不用选择是DNA 还是蛋白质序列比对,选择的也是fasta格式的文件,打开后的界面都是一样的。
以第一种情况为例说明。
点击如出现下界面:这里我们分析的是蛋白序列所以选择No。
点击出现如下界面:然后从data菜单选择输入数据文件如图:选择你保存的fasta格式序列后就会出现:下面介绍菜单的使用:Data:Creat a new :创建一个新的数据比对文件,也就是说当我们比对完一组后,想接着比对另一组,那么使用它就可以不用退出直接把数据文件导入;Open:打开先前已经比对并保存好的文件,它包含两个子菜单:retive sequence from file和saved aligment session ;Close: 关闭当前的比对数据文件;Save session:保存当前比对结果,可以给比对的结果一个文件名;Export alignment:将当前的序列比对结果输出到指定文件,有两种输入格式可供选择:MGTA和FASTA.DNA sequence:使用它来选择输入的数据DNA序列,这里需要说明的是如果你输入的数据是氨基酸序列的话,比对窗口只显示一个标签,若是DNA序列的话则显示两个标签,一个是DNA序列的,另一个是氨基酸序列的。
如图:Protein sequences:选择输入的氨基酸序列,选择后,所以的位点就被当作氨基酸残基位点来对待。
Translate/untranslate:只有比对的序列是编码蛋白的DNA序列的时候才可用。
它可以根据指定的遗传密码表将DNA序列翻译成特定的氨基酸序列。
Select genetic code table:使用它将编码蛋白的DNA翻译成特定的蛋白序列。
Reverse complement:将选择的一整行的DNA序列变为与之互补配对碱基序列。
Exit alignment explorer:退出序列比对的资源管理窗口。
Edit菜单:使用这个菜单可以对我们的比对序列进行想要的一些编辑工作具体为Undo:撤销上一步操作;Copy:复制;cut:剪切;Paste:粘贴;前面三个操作都可以只针对一个碱基或氨基酸残基也可以是一段甚至是整个序列;Delete:从比对表格中删除一段序列;Delete gaps:去掉序列中的空缺;Insert blank sequence:重新插入一空行;标签和序列都是空的;Insert sequence from file:从已保存的文件中插入新的序列;Select sites:选择一列序列,与点击比对表上方的灰白空格作用类似;Select sequence:选择一行序列,与点击比对表格左侧的标签名作用类似;Select all:全选;Allow base editing:只读保护,只有选择后才能对序列进行编辑操作,否则所以的序列为只读格式,不能进行任何编辑操作。
Search菜单:用来快捷查找序列中的标记未定或者目的碱基或残基。
Find motif:选择后出现如下对话框:输入你想要查看的一小段序列。
找到后会以黄色标出;Find next:在序列的下游查找目的序列片段;Find preious:在序列的上有查找目的序列片段;Find marked sites:查找标记位点;Highlight motif:突出标记已经选择的位点。
Web菜单这个菜单提供一个链接Genbank的入口,可以在网上直接做Blast搜索。
当手上没有准备好要比对的序列时,可以直接去网上搜索。
Query gene banks:开启NCBI的主页;Do blast search:开启NCBI BLAST主页;Show browser:开启网页浏览器。
Sequencer菜单此菜单下只有一个子菜单:edit sequencer file,用来打开一个打开文件对话框,此对话框可以打开一个sequencer data file,一旦打开,这个文件就在trace data file viewer/editor的对话框中展示出来。
这个编辑窗口允许你查看和编辑automatd DNA sequencer 产生的trace data。
它可以阅读和编辑ABI和Staden格式文件并且序列可以直接被导入到序列比对窗口或被上传到网页浏览器做blast搜索。
Display菜单:这个菜单相对简单,主要用来调整工具栏。
Toolbars:工具栏菜单,它包含一些子菜单,选择后就会出现在比对的窗口中;Use colors:将不同的位点以不同的颜色显示;Background color:选择后位点的显示与位点一样的背景颜色;Font:字体对话框,通过选择来调整窗口中的序列字符的大小。
最后,结合实例来介绍alignment 菜单Mark/unmark site:在比对的表格中标记或者不标记一个单一位点,一次每条序列只能被标记一个位点,不同序列间的位点你可以选择同一列的,也可以是错开的,要根据自己的目的进行选择。
选择标记后的序列可以使用align marked sites 进行比对分析。
Align marked sites:比对标记的序列,在这里如果在两个或多个序列间标记了不在一列的位点重新比对后会出现空格。
如图:Unmarked all sites:把所以标记的位点去标记;Delete gap-only site:去掉序同是空格的一列;这在多序列比对前很有用。
Auto-fill gaps:使用空格补齐不同长度的序列。
Align by clustalw:此软件整合了clustalw程序,这也是它的方便之处,选择要比对的序列后点击会出现下面的对话框:一般参数:DNA/protein weight matrix:选择不同的加权矩阵;Residue-specific penalties:特殊氨基酸罚分。
在序列比对的过程中特异氨基酸可能增加或减少罚分值,比如:富含甘氨酸的区段比富含缬氨酸的区段更可能有空格出现,因而他们的罚分不同。
Hydrophilic penalties:如果有连续的5个或者更多的亲水性氨基酸的话,他们倾向于出现空格,这些区段很可能出现环状或卷曲,因此罚分不一样。
Gap separation distance:参数设置来尽可能降低空格之间离的太近的机会,小于指定数值的空格罚分要多余其他的,这不能避免出现相邻空格,只能降低他们出现的频率。
Use negative matrix:使用负性矩阵,Delay divergent cutoff:若一条序列相似性低于设定的百分值将推迟比对。
当一切参数都设定好了之后就点击OK就可以进行比对了,中间出现一个过度对话框,等一下就出现如下界面:注意:以上介绍的许多操作菜单在窗口的上放有快捷操作按钮。
如上图标记内。
比对结束后,可以将结果保存(data/save session/),以供构建系统发育树使用。
另外,如果不保存直接关闭,系统跳出一个对话框:下面这个是序列数据管理的管理界面,此外我们还可以通过主界面上的data/open data路径打开,效果是一样的,注意这里打开的只能是刚才保存的后缀是.MEG的文件。
几个功能菜单:下面就着重介绍一下序列数据窗口的一些具体使用:这个窗口用来展示比对后的序列数据,这里提供了许多的功能菜单用来查看序列比对后的数据统计结果或者来选择想要的子序列。
Data 菜单Write data to file:导入序列打开窗口;Translate/untranslate:将蛋白编码序列翻译成蛋白序列,或者再转变成核酸序列;Selected genetic code table:打开select genetic code对话框,从这个对话框可以选择编辑或者添加遗传密码表;Setup/select genes and domains:打开sequence data organizer对话框,在这个对话框里可以定义和编辑基因和结构域。
使用这个窗口可以查看,定义,和选择结构域和基因,并且标记单个的位点。
具体使用这里不作详细介绍。
Setup/select taxa and groups:打开一个可以编辑分类和定义分类组的对话框:这个窗口分为两个子窗口,左边的是分类组,显示不同的分组情况,右边的是未分组窗口显示还没有归入任何一个组群的分类。
中间和下边是一些操作键,通过他们我们可以建立新的组,如果你将所以的分类都归入到不同的组里,并且给予组名,你们在序列数据窗口中就会在物种名字后边显示他所属的组名。
Quite data viewer:退出界面。
Display 菜单:Show only selected sequence:只显示你所选择的感兴趣的序列;Use identical symbol:将一列中绝大部分相同等碱基或氨基酸字符用点来代替;Color cells:将序列中连续的一致的碱基或者氨基酸给以相同的颜色背景以区别显示;Sort sequences:将显示的分类以不同的方式排序,可以根据序列名字、组名来排序;Restore input order:将经过修改的序列顺序回复到刚打开时的样子;Show sequence name:显示序列的名字,不选则隐藏;Grouped:显示组名;Change font:更改显示的字体格式。