MEGA软件的使用
- 格式:doc
- 大小:1.39 MB
- 文档页数:13

Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。
在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息。
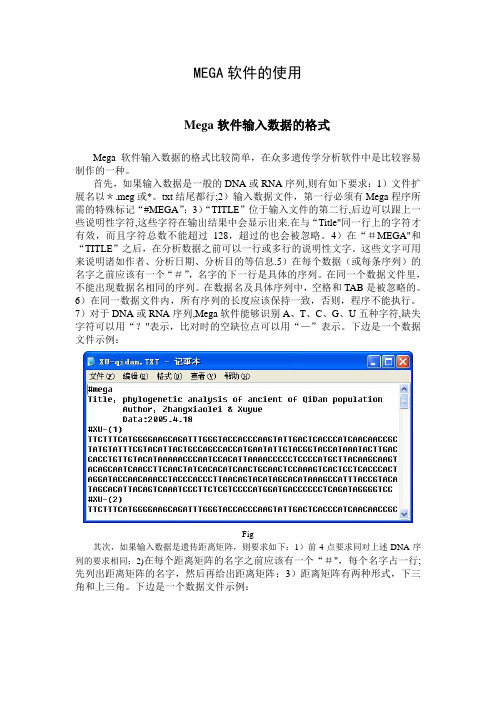
5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:Fig下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
Fig再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
MEGA软件的使用Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*。
txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来.在与“Title"同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA"和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息.5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?"表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#",每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:Fig下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
Fig再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等.而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
MEGA软件的使用Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。
在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息。
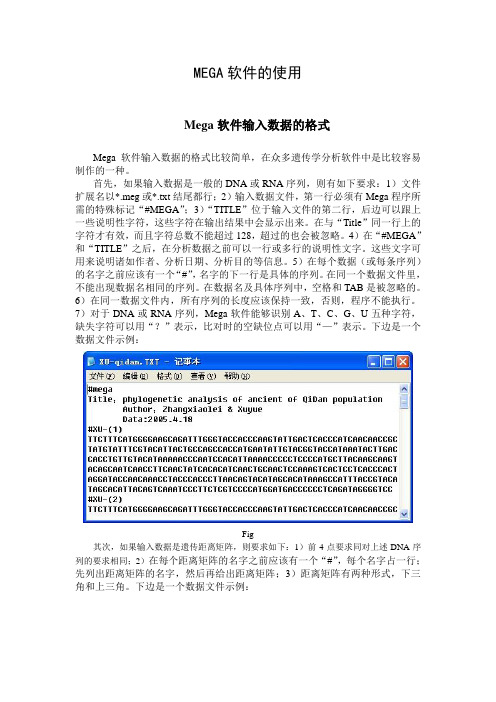
5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
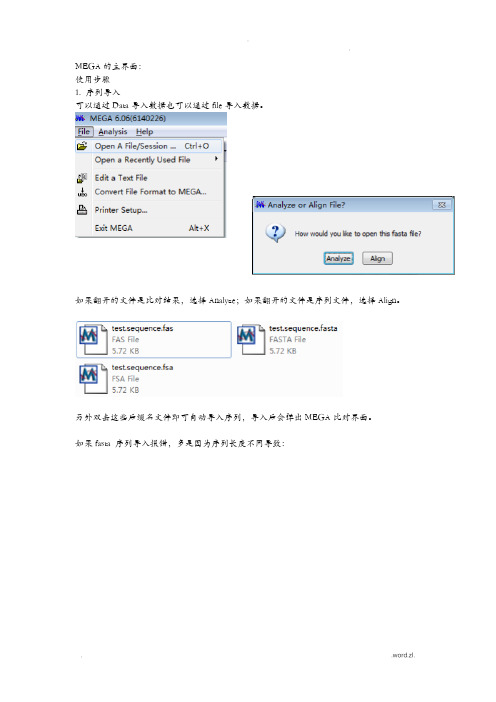
MEGA的主界面:使用步骤1. 序列导入可以通过Data导入数据也可以通过file导入数据。
如果翻开的文件是比对结果,选择Analyze;如果翻开的文件是序列文件,选择Align。
另外双击这些后缀名文件即可自动导入序列,导入后会弹出MEGA比对界面。
如果fasta 序列导入报错,多是因为序列长度不同导致:如果序列长度不同,可以采用新建文件,将序列文件导入的方法。
步骤:Align →Edit/Build Alignment →create a new alignment →Data →open →Retrieve sequences from File将复制输入的序列另存输出看看。
步骤:data →Export alignment →fasta format序列长度都被用横线补齐了。
2. 多序列比对选择muscle或者clustalw进展比对:clustalw 一般用于DNA ,muscle多用于蛋白。
在比对之前需先选中要进展比对的序列〔Shift〕,还可以对序列或者序列名进展编辑〔双击〕。
比对参数选择:保存比对文件,进化树分析提供数据。
一般导出的比对结果保存为fasta格式,或者直接点击保存按钮将结果,保存为二进制的mas 或meg文件。
3. 构建进化树导入数据:将刚刚另存的meg 文件重新导入到mega程序中〔直接拖入工作界面〕,并选择构建进化树。
参数选择:参数设置,Bootstrap method一般选择1000~1500;第一次绘图时建议选择500,这样运行速度会比价快,结果适宜再调至1000重新进展进化分析。
描述:进化树可视化:View→Tree/Branch style选择树的模式,也可以通过右图菜单进展选择。
发散树环状树进化树简单美化:可视化文件的保存:•Newick——标准树文件,用于下游可视化软件的导入•png——压缩图像文件•pdf——矢量图像文件保存为png/pdf,显示不全怎么办?——将图和图注复制到WORD中保存即可〔Image Copy to Clipboard 粘贴到WORD〕。
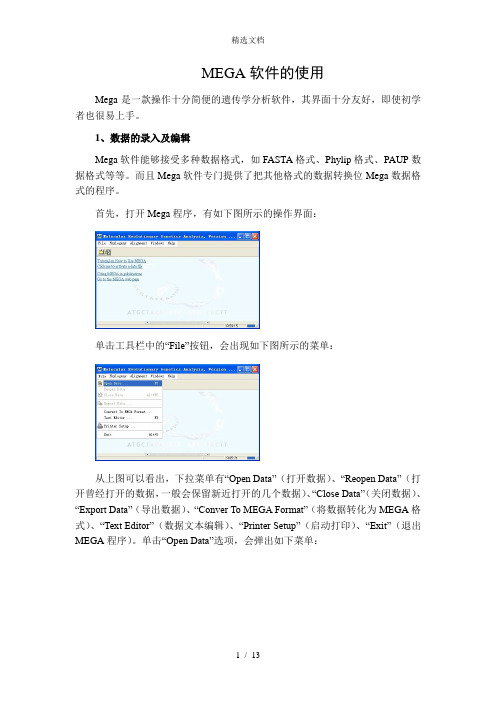
MEGA软件的使用Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,打开Mega程序,有如下图所示的操作界面:单击工具栏中的“File”按钮,会出现如下图所示的菜单:从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。
单击“Open Data”选项,会弹出如下菜单:浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面:此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。
根据输入数据的类型,选择一种,点击“OK”即可。
如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示:根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。
点击“OK”按钮,即可导入数据。
如果是核苷酸数据,则读完之后,会弹出如下对话框:如上图,如果是编码蛋白质的核苷酸序列,则选择“Yes”按钮;如果是不编码蛋白质的核苷酸序列,则点击“No”按钮。
之后,会弹出如下操作窗口:此作界面的名称是“Sequence Data Explorer”,在其最上方是工具栏“Data”、“Display”、“Highlight”等,然后是一些数据处理方式的快捷按钮,在操作界面的左下方是每个序列的名称。

MEGA软件构建系统发育树摘要:以白色念珠菌属下面的十个种的18s RNA 为例,构建系统发育树来说明MEGA 软件的使用方法。
1背景简介1.1 MEGA(分子进化遗传分析)MEGA 的全称是Molecular Evolutionary Genetics Analysis。
MEGA is an integrated tool for automatic and manual sequence alignment, inferring phylogenetic trees, mining web-based databases, estimating rates of molecular evolution, and testing evolutionary hypotheses. MEGA 可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说等。
MEGA 还可以通过网络(NCBI)进行序列的比对和数据的搜索。
最新版本:MEGA 5.1 Beta (软件开发者建议其结果不用于发表文章)建议下载版本:MEGA 5.05 for Windows and Mac OS。
MEGA 5 has been tested on the following Microsoft Windows® operating systems: Windows 95/98, NT, 2000, XP, Vista, version 7, Linux and Mac OS [1].MEGA 5.05 可免费下载,只需输入名字及有效邮箱,下载链接会发送至邮箱,点击可下载。
1.2 系统发育树定义系统发育树(英文:Phylogenetic tree)又称为演化树(evolutionary tree),是表明被认为具有共同祖先的各物种间演化关系的树。
是一种亲缘分支分类方法(cladogram)。
在树中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)1.3 系统发育树的分类根据有根和无根来区分:树可分为有根树和无根树两类。
MEGA软件的使用Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,打开Mega程序,有如下图所示的操作界面:单击工具栏中的“File”按钮,会出现如下图所示的菜单:从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。
单击“Open Data”选项,会弹出如下菜单:浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面:此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。
根据输入数据的类型,选择一种,点击“OK”即可。
如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示:根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。
点击“OK”按钮,即可导入数据。
如果是核苷酸数据,则读完之后,会弹出如下对话框:如上图,如果是编码蛋白质的核苷酸序列,则选择“Yes”按钮;如果是不编码蛋白质的核苷酸序列,则点击“No”按钮。
之后,会弹出如下操作窗口:此作界面的名称是“Sequence Data Explorer”,在其最上方是工具栏“Data”、“Display”、“Highlight”等,然后是一些数据处理方式的快捷按钮,在操作界面的左下方是每个序列的名称。
MEGA软件的使用MEGA软件的使用Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP 数据格式等等。
而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,翻开Mega程序,有如以下图所示的操作界面:单击工具栏中的“File〞按钮,会出现如以下图所示的菜单:从上图可以看出,下拉菜单有“Open Data〞〔翻开数据〕、“Reopen Data〞〔翻开曾经翻开的数据,一般会保存新近翻开的几个数据〕、“Close Data〞〔关闭数据〕、“Export Data〞〔导出数据〕、“Conver To MEGA Format〞〔将数据转化为MEGA格式〕、“Text Editor〞〔数据文本编辑〕、“Printer Setup〞〔启动打印〕、“Exit〞〔退出MEGA程序〕。
单击“Open Data〞选项,会弹出如下菜单:浏览文件,选择要分析的数据翻开,单击“翻开〞按钮,会弹出如下操作界面:此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences〔核苷酸序列〕、Protein Sequences〔蛋白质序列〕、Pairwise Distance〔遗传距离矩阵〕。
根据输入数据的类型,选择一种,点击“OK〞即可。
如果选择“Pairwise Distance〞,那么操作界面有所不同;如以下图所示:根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix〞即可;如果是上三角矩阵,选择“Upper Right Matrix〞即可。
点击“OK〞按钮,即可导入数据。
如果是核苷酸数据,那么读完之后,会弹出如下对话框:如上图,如果是编码蛋白质的核苷酸序列,那么选择“Yes〞按钮;如果是不编码蛋白质的核苷酸序列,那么点击“No〞按钮。
MEGA软件的使用Mega软件输入数据的格式Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。
在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。
4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。
这些文字可用来说明诸如作者、分析日期、分析目的等信息。
5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。
在同一个数据文件里,不能出现数据名相同的序列。
在数据名及具体序列中,空格和TAB是被忽略的。
6)在同一数据文件,所有序列的长度应该保持一致,否则,程序不能执行。
7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。
下边是一个数据文件示例:Fig其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。
下边是一个数据文件示例:Fig下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
Fig再次,如果数据是测序图谱的形式,直接导入即可。
下图是测序图谱示例:FigMEGA界面及操作Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5首先打开MEGA 5软件,界面如下:然后,导入需要构建系统进化树的序列:点击OK出现新的对话框,创建新的数据文件导入成功3)序列比对分析点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建以NJ为例Bootstrap选择1000,点Computer,开始计算计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法5)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
MEGA软件的使用Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。
而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,打开Mega程序,有如下图所示的操作界面:单击工具栏中的“File”按钮,会出现如下图所示的菜单:从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。
单击“Open Data”选项,会弹出如下菜单:浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面:此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。
根据输入数据的类型,选择一种,点击“OK”即可。
如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示:根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。
点击“OK”按钮,即可导入数据。
如果是核苷酸数据,则读完之后,会弹出如下对话框:如上图,如果是编码蛋白质的核苷酸序列,则选择“Yes”按钮;如果是不编码蛋白质的核苷酸序列,则点击“No”按钮。
之后,会弹出如下操作窗口:此作界面的名称是“Sequence Data Explorer”,在其最上方是工具栏“Data”、“Display”、“Highlight”等,然后是一些数据处理方式的快捷按钮,在操作界面的左下方是每个序列的名称。
显示序列占了操作界面的绝大部分,与第一个序列相同的核苷酸用“.”表示,发生变异的序列则直接显示。
2、遗传距离的计算点击Mega操作主界面的“Distances”按钮,会弹出一个下拉菜单。
如下图所示:从上图易知,此菜单包括如下选项:“Choose Model”(选择模型,即选择计算遗传距离的模型)、“Compute Pairwise”(计算遗传配对差异)、“Compute OverallMean”(计算包括所有样本在内的平均遗传距离)、“Compute With Group Means”(计算组内平均遗传距离)、“Compute Between Groups Means”(计算组间平均遗传距离)、“Compute Net Between Groups Means”(计算组间平均净遗传距离)、“Compute Sequence Diversity”(计算序列分歧度)。
“Compute Sequence Diversity”选项包括四个子菜单:“Mean Diversity Within Subpopulations”(亚群体内部平均序列多态性)、“Mean Diversity for Entire Population”(整个人群平均序列多态性)、“Mean Interpopulaional Diversity”(群体内部平均序列多态性)、“Coefficient of Differentiation”(遗传变异系数)。
点击“Choose Model”选项,会弹出如下操作界面:从上述操作界面可以看出,通过此对话框可以选择计算遗传距离的模型等。
“Data Type”显示数据的类型:Nucleotide(Coding)(编码蛋白质的DNA序列)、Nucleotide(不编码蛋白质的DNA序列)、Amino Acid(氨基酸序列)。
通过“Model”选项可以选择,计算遗传距离的距离模型。
点击“Model”一行末端的按钮会弹出一选择栏。
如上图所示,对于非编码的核苷酸序列Mega程序提供了八种距离模型:“Number of Difference”(核苷酸差异数)、“P-distance”(P距离模型)、“Jukes-Cantor”(Jukes和Cantor距离模型)、“Kimura 2-Parameter”(Kimura双参数模型)、“Tajima-Nei”(Tajima和Nei距离模型)、“Tamura 3-parameter”(Tamura 三参数模型)、“Tamura-Nei”(Tamura和Nei距离模型)、“LogDet(Tamura kumar)”(对数行列式距离模型)。
对于编码的核苷酸序列,其遗传距离模型如下图所示:如上图所示,对于编码蛋白质的DNA序列,Mega程序提供了一下几种模型:“Nei-Gojobori Method”,“Modified Nei-Gojobori Methoed”、“Li-Wu-Luo Method”、“Pamilo-Bianchi-Li Method”、“Kumar Method”。
其中Nei-Gojobori方法和修正的Nei-Gojobori方法都包含三种距离模型:“Number of Differences”、“P-distance”、“Jukes-Cantor”。
对于氨基酸序列,Mega所提供的遗传距离模型如下图所示:如上图所示,对于氨基酸序列,Mega程序提供了一下六种遗传距离模型:“Number of Differences”(氨基酸差异数)、“P-distance”(P距离模型)、“Poisson Correction”(泊松校正距离模型)、“Equal Input”(等量输入距离模型)、“PAM Matrix(Dayhoff)”(PAM距离矩阵模型)、“JTT Matrix(Jones-Taylor-Thornton)”(JTT距离矩阵模型)。
在“Analysis Preference”操作界面中,“Pattern Among Lineages”仅提供了一个选项:“Same(Homogenous)”“,也就是说样本之间是有一定同源性的。
“Rates among sites”提供了两个选项:“Uniform Rates”和“Different(Gamma Distributed)”。
“Uniform Rates”意味着所有序列的所有位点的进化速率是相同的。
选择“Different (Gamma Distributed)”,意味着序列位点之间的进化速率是不相同的,可以利用Gamma参数来校正,系统提供了四个数值可供选择:2.0、1.0、0.5、0.25;软件使用者也可以自行决定Gamma参数的大小。
设置完毕后,在此界面中点击“OK”按钮,即可返回Mega操作主界面。
选择主操作界面“Distance”中的“Compute Pairwise”选项,可以计算样本之间的遗传距离的大小,其操作界面如下图所示:“Data Type”显示数据的类型,图中为“Nucleotide”。
“Analysis”显示计算分分析的类型,图中为“Pairwise Distance Calculation”(配对差异距离计算)。
“Compute”显示所要运行的对象,又两个选项:“Distance only”(仅计算遗传距离)和“Distance&Std.Err”(计算遗传距离和其标准误)。
“Include Sites”显示利用哪些位点来计算,如果数据类型是不编码蛋白质的核苷酸序列,则全部参与计算,如果是编码蛋白质的核苷酸序列,则可以选择哪些位点(如密码子的第2位等)来参与运算。
“Substitution Model”是替代的模型,在下边“Model”中可以进行选择。
“Substitutions to Inclued”选择哪些替代类型(如下图所示)被用于运算,d 选项将转换和颠换全部包括在内,s选项仅包括转换,v选项仅包括颠换,R为转换和颠换的比值,L为所有有效的普通位点的个数。
“Pattern among Lineages”和“Rates among sites”上文已有介绍,不再详述。
点击“Compute”按钮,即可开始计算。
其显示运算结果的界面如下图所示:上图是计算出的各个样本之间的遗传距离的矩阵。
在最下端的状态栏,显示的是所利用的遗传距离模型,如图中所示:Nucleotide:Kimura 2-parameter。
“File”按钮共有四个下拉菜单:“Show Input Data Title”(显示输入数据的标题)、“Show Analysis Description”(显示分析信息的描述)、“Export/Print Distance”(输出或打印距离矩阵)、“Quit viewer”(退出此操作界面)。
“Display”按钮共有四个下拉菜单:“Show Pair Name”(显示配对序列的名字)、“Sort Sequence”(用何种方式对序列进行排序)、“Show Names”(显示序列的名字)、“Change Font”(改变字体)。
“Sort Sequence”有两个选项:“Original”(按原先输入的顺序)和“By Name”(通过序列的名字)。
点击“Average”按钮可以计算平均的遗传距离,此按钮提供了四个下拉菜单:“Overall”(所有样本之间的平均遗传距离)、“Within Groups”(组内平均遗传距离)、“Between Groups”(组间平均遗传距离)、“Net Between Groups”(组间平均净遗传距离)。
在上述按钮下方还有六个按钮,如下图所示。
点击第一个按钮可以使数据以下三角矩阵的方式显示;点击第二个按钮可以使数据以上三角矩阵的方式显示;选中第三个按钮可以显示配对的序列的名字,点击第四个按钮,可以减少数据小数点后的位数;点击第五个按钮,可以增加数据小数点后的位数;拖动第六个按钮中的小竖条可以改变数据显示的宽度。
点击“File”下拉菜单中的“Export/Print Distance”选项,会弹出如下图所示的对话框:“Output Format”选项可以确定输出数据的格式:“Publication”(一般格式)和“Mega”(Mega格式,把此数据保存可直接由Mega程序打开,进行构建系统发育书等遗传分析)。
Decimal Places(小数位的大小),“Max Entries per line”(每一行最多能显示的数据的个数)。