常染色体显性多囊肾病研究现状及进展
- 格式:pdf
- 大小:331.17 KB
- 文档页数:5

《中国产前诊断杂志(电子版)》 2020年第12卷第1期·综述· 胎儿常染色体显性遗传性多囊肾病的研究进展朱雨露 综述 徐岚 审校(汕头大学医学院第一附属医院,广东汕头 515041)【摘要】 常染色体显性多囊肾病(autosomal dominantpolycystickidneydisease,ADPKD)是最常见的遗传性肾病之一。
通常,ADPKD在早期无明显自觉症状,多在成年后发病,临床上该病以双肾多发性囊肿为主要特征,并常伴发肝、胰、脾等器官囊性病变及心脑血管病变。
ADPKD亦可在儿童期甚至在胎儿期发病,一旦发病,其程度较成年患者更加严重,疾病进展也更加迅猛。
诊断为患有ADPKD的胎儿的母亲在随后的妊娠中,胎儿有50%的复发机会。
胎儿超声检查在胎儿常染色体显性多囊肾病的病因学研究和预后评估中是必不可少的,在症状出现前,产前即进行胎儿基因检测并尽早干预有助于避免具有致命缺陷的胎儿出生,目前已经有多种基因测试方法可用于检测ADPKD。
本文旨在对胎儿常染色体显性遗传性多囊肾病在发病机制、临床特点、产前检查、产前诊断与治疗方面的最新研究进展进行综合评述。
【关键词】 遗传咨询;先天畸形;产前诊断;产前超声;常染色体显性遗传性多囊肾病;遗传学【中图分类号】 R714.55 【文献标识码】 A犇犗犐:10.13470/j.cnki.cjpd.2020.01.012 通信作者:徐岚,E mail:xulandoctor@163.com 常染色体显性多囊肾病(autosomal dominantpolycystickidneydisease,ADPKD)是一种以常染色体显性遗传方式遗传的疾病,是一种累及多系统、多脏器的全身性疾病。
在活产婴儿中发病率约为1/500~1/1000[1],大约8%的透析或接受肾移植的患者患有ADPKD[2]。
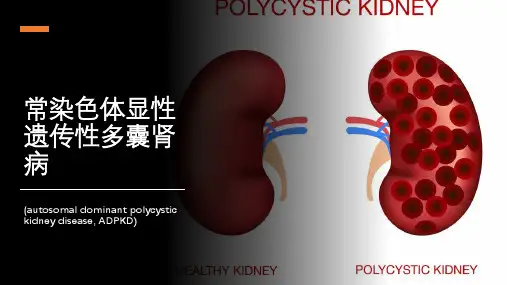
ADPKD的特征在于双肾多发性囊肿,该疾病的标志是在儿童早期肾脏中数百个微观充满液体的囊肿的发展,它们以不同的速率甚至以指数倍的方式连续生长(每年增加2%~10%),逐渐导致正常肾组织的损失[3],最终将导致终末期肾病(end stagekidneydisease,ESKD)。



常染色体显性遗传多囊肾研究进展摘要常染色体显性遗传多囊肾(ADPKD)是一种发病率高、预后差的疾病,它在发病机制、治疗等方面有很多进展。
关键词常染色体显性遗传多囊肾;瞬时受体势;Max作用因子1;雷帕霉素靶蛋白常染色体显性遗传多囊肾(autosomal dominant polycystic kidney disease,ADPKD)是一种常见的遗传性肾病,是导致肾衰竭的重要疾病。
现在已经发现3个基因(PKD1、PKD2、PKD3)与此病有关,其中PKD1定位于染色体16p13.3,其突变而导致ADPKD约占85%,PKD2定位于染色体4q21-23,其突变约占15%。
PKD3突变仅在几个家族中发现,目前尚未定位[1]。
ADPKD病变以双肾多发性进行性充液囊泡为主要特征。
囊泡损伤肾组织,引起肾功能改变,出现血尿、蛋白尿等临床症状,最终导致肾衰竭。
ADPKD除累及肾脏外,还可引起肝脏囊肿、胰腺囊肿、心脏瓣膜病、结肠憩室和颅内动脉瘤等肾外病变[2],给患者、家属及社会带来沉重负担。
故揭示其发病机理,研究新的治疗方法有很重要的意义。
本文对这些进展作以综述。
1 ADPKD的发病机制1. 1 多囊蛋白PC PKD1基因的蛋白产物被称为多囊蛋白-1(polycystin-1,PC1),又叫做TRPP1,多囊蛋白-1是一种跨膜蛋白,分布广泛,可以与多种蛋白(如PC2)、糖、脂类结合并发生交互作用,从而发挥功能。
PC1还与Wnt信号途径、JAK-STAT途径、转录因子AP-1和G蛋白偶联等信号途径有关。
PKD2基因的蛋白产物被称为多囊蛋白-2(polycystin-2,PC2),又叫做TRPP2,是TRP家族的一员,为非选择性钙离子通道。
它不同于PKD1,在人类基因组中是单拷贝,并不含多嘧啶区,但它的第一个外显子富含GC,从而使得此处易于突变[3]。
瞬时受体势(transient receptor potential,TRP)通道虽然最初发现于感受器,主要参与神经传导,但随着研究的深入,近年来人们发现该通道在肾脏病的发生、发展中也发挥了作用。


常染色体显性遗传病之多囊肾病的研究和治疗摘要:控制一种遗传性状的显性基因位于常染色体上,其遗传方式称为常染色体显性遗传(AD),由这种致病基因引起的疾病称为常染色体显性遗传病。
常染色体显性遗传型多囊肾病(autosomal Dominant polycystic kidney disease,ADPKD)是一种最常见的单基因遗传性肾病,发病率约为1/400~1/1000。
ADPKD多在3()岁~50岁之间发病,囚此过去常称为“成人型多囊肾病”,实际上ADPKD可在任何年龄发病,包括妊娠时的胎儿,故“成人型”这一术涪并不准确,现已废用。
ADPKD临床表现为双侧肾脏皮、髓质有多个液性囊肿形成和增大,晚期伴肾功能损害。
可累及多个系统。
正文:一多囊肾病的概述1.什么叫多囊肾多囊肾是肾脏的皮质和髓质出现多个囊肿的一种遗传性肾脏疾病。
2.还患者发病原因90%异常基因位于16号染色体的短臂。
另有10%不到患者的异常基因位于4号染色体的短臂。
患者的基因型在常染色体显性遗传病中,假定用A表示显性致病基因,a表示相对应的隐性正常基因,则患者的基因型有两种,显性纯合体(AA)和杂合体(Aa),基因型aa的个体正常,但临床上所见到的患者大多数为杂合体。
二.系谱特点1、男女发病几率相等;2、父母有一方患病,子女有50%获得囊肿基因而发病,如父母均患此病,子女发病率增加到75%;3、不患病的子女不携带囊肿基因,其下代(孙代)也不会发病,即不会隔代遗传。
真正不经父母遗传而由基因突变而发病的情况极少见。
三.多囊肾的症状1、肾肿大两侧肾病变进展不对称,大小有差异,至晚期两肾可占满整个腹腔,肾表面布有很多囊肿,使肾形不规则,凹凸不平,质地较硬。
2、肾区疼痛为其重要症状常为腰背部压迫感或钝痛,也有剧痛,有时为腹痛。
3、血尿约半数病人呈镜下血尿,可有发作性肉眼血尿,此系囊肿壁血管破裂所致。
4、高血压。
5、肾功能不全本病迟早要发生肾功能不全。

美国多囊肾最新研究报告美国多囊肾最新研究报告引言多囊肾是一种常见的肾脏遗传性疾病,其特征是肾脏内出现多囊状扩张。
这种疾病通常会导致慢性肾功能衰竭,对患者的生活质量和寿命都有严重影响。
近年来,美国的研究人员在多囊肾的治疗和预防方面做出了重要的突破,本文将对美国多囊肾最新的研究进展进行综述。
1. 多囊肾的病因和遗传机制多囊肾是一种常染色体显性遗传的疾病,主要由PKD1和PKD2基因突变引起。
这两个基因编码了多囊肾蛋白1和多囊肾蛋白2,这两种蛋白在肾小管上皮细胞中起着重要的调控作用。
突变导致了多囊肾蛋白的功能失调,进而导致肾小管细胞功能障碍和增生。
2. 分子机制研究的进展近年来,美国的研究人员通过对多囊肾基因的研究,揭示了多囊肾发病的分子机制。
他们发现,多囊肾蛋白在细胞内形成复合物,并与其他蛋白相互作用,调控细胞增殖和分化。
同时,他们还发现了多囊肾蛋白对一些细胞信号通路的调节作用。
这些研究结果为多囊肾的治疗提供了新的靶点和方向。
3. 研究进展近年来,美国的研究人员在多囊肾的治疗和预防方面做出了重要的突破。
他们发现了一些潜在的药物和干预方法,可以延缓疾病的进展,并改善患者的生活质量。
以下是一些相关的研究进展:•药物治疗–研究人员发现一些已经上市的药物对多囊肾有一定的治疗作用,例如利拉鲁肽。
–研究人员还通过筛选化合物库发现了一种新的药物,具有降低多囊肾蛋白表达的作用。
•细胞治疗–美国的研究人员利用干细胞技术,成功地将修复后的肾小管细胞移植到多囊肾患者的肾脏中,取得了显著的治疗效果。
–另外,研究人员还通过基因编辑技术,成功地修复了多囊肾基因突变,为多囊肾的基因治疗提供了新的思路。
•遗传咨询和干预–研究人员提出了遗传咨询和干预的重要性,通过遗传咨询,患者和家族成员可以了解遗传风险,并采取相应的预防措施。
–研究人员还通过基因编辑技术,成功地修复了多囊肾基因突变,为多囊肾的基因治疗提供了新的思路。
4. 未来的研究方向美国的多囊肾研究在治疗和预防方面取得了重要的进展,但仍有许多问题需要解决。



[基金项目]山东省自然科学基金资助项目(Y 2002C 12)。
・综述与讲座・常染色体显性多囊肾基因突变研究进展刘 征,丁克家(山东省立医院,山东济南250021)[关键词] 遗传性疾病;多囊肾疾病;基因突变[中图分类号] R 69211 [文献标识码] A [文章编号] 10022266X (2006)2120090202 常染色体显性多囊肾病(AD PKD ),又称成人型多囊肾病。
据国外文献报道,到60岁时大约有50%的患者出现终末期肾衰竭。
该病为单基因遗传病,存在遗传异质性。
目前已知的引起该病的突变基因可能有3个,分别为PKD 1基因、PKD 2基因、PKD 3基因。
其中85%患者由PKD 1基因突变引起[1],约15%患者由PKD 2基因突变引起。
PKD 3基因由于少见,研究不多,故至今尚未定位克隆。
1 PKD 1基因PKD 1基因定位在第16号染色体短臂1区3带3亚带(16p 1313),总跨度约53kb ,由46个外显子组成,其转录的mRNA 长度1411kb ,编码的多囊蛋白产物称为PC 1。
PKD 1基因的近端区域(16p 1311)含有三个同源基因位点(H G 2A 、H G 2B 和H G 2C ),与PKD 1基因同源性极高。
PKD 1基因的35~46外显子为单拷贝区,1~34外显子由于存在同源基因出现3次核苷酸重复,这给基因测序和突变检测带来了极大困难。
为此,不同实验室采用了多种不同方法,包括蛋白截断实验(PT T )、变性梯度凝胶电泳(D GGE )、单链构象多态性分析(SSCP )、高压液相色谱法(DH PL C )及直接测序法。
每种方法各有利弊,PT T 无法检测没有造成蛋白异常终止的错义突变;D GGE 由于需要在电泳前在产物两次加上一对GC “夹子”,增加了操作难度;SSCP 虽然简便,但存在5%~30%的漏检率;DH PL C 需要专门的仪器设备;直接测序虽然检出率较高,但花费较大。
多囊卵巢综合征在中国的诊治现状和研究进展1. 引言1.1 多囊卵巢综合征在中国的诊治现状多囊卵巢综合征是一种常见的内分泌紊乱性疾病,患者主要表现为月经紊乱、排卵障碍、多囊卵巢等症状。
根据国内相关数据显示,多囊卵巢综合征在中国女性中的患病率逐年增加,已成为重要的妇科问题之一。
在中国,多囊卵巢综合征的诊治现状存在一些问题。
首先,部分患者对疾病的认识不足,导致延误就诊。
其次,由于临床症状多样化,很多医生对多囊卵巢综合征的诊断与治疗仍存在争议和困惑。
另外,由于病因未明,目前尚无特效的治疗方法,治疗效果参差不齐。
此外,部分地区医疗资源分布不均,导致患者就医困难,影响了疾病的有效管理。
尽管存在种种挑战,但在中国,相关专家学者和医疗机构正努力改善多囊卵巢综合征的诊治现状。
通过加强宣传教育,提高医疗服务水平,推动多囊卵巢综合征的诊疗规范化,相信未来在中国的多囊卵巢综合征诊治中会取得更好的成果。
1.2 多囊卵巢综合征在中国的研究进展多囊卵巢综合征是一种常见的内分泌失调性疾病,近年来在中国的研究进展取得了一些重要成果。
通过大量的流行病学调查和临床研究,中国学者已经对多囊卵巢综合征在中国的发病率和特点有了更深入的了解。
据中国医学研究显示,多囊卵巢综合征的患病率已经呈现逐年上升的趋势,尤其是在城市女性中更为普遍。
中国的科研人员在多囊卵巢综合征的病因和发病机制方面进行了大量的研究。
他们发现,遗传、环境、生活方式等诸多因素都可能对多囊卵巢综合征的发生起到一定的作用,从而为今后更深入的病因探讨奠定了基础。
中国在多囊卵巢综合征的治疗方法和药物研究方面也取得了一些进展。
包括中药治疗、针灸、运动疗法等在内的多种治疗手段都得到了一定程度的验证,为临床治疗提供了更多的选择。
中国在多囊卵巢综合征的研究进展中正逐渐走向多元化和深入化的方向,未来还有待进一步的研究和探讨。
2. 正文2.1 多囊卵巢综合征的临床表现和诊断方法多囊卵巢综合征是一种常见的内分泌失调疾病,其临床表现多种多样,主要包括月经紊乱、不孕不育、多毛症、肥胖等。
托伐普坦治疗常染色体显性多囊性肾病患者的研究进展刘玉法;陈广新;刘纳新;苏立军【期刊名称】《中国生化药物杂志》【年(卷),期】2014(000)007【摘要】通过搜索PubMed,ScienceDirect,万方,CNKI数据库,选择1990~2014年文献,对托伐普坦治疗常染色体显性多囊性肾病(autosomal dominant polycystic kidney disease,ADPKD)研究情况进行综述、总结。
常染色体显性多囊肾病是一种慢性进行性疾病,可显著地增加经济负担和致死率。
目前尚没有特定药物用于治疗或延缓该病的进程。
ADPKD患者早期使用托伐普坦可减慢疾病进程,但可能伴发频繁的、严重的不良事件,需密切监护。
%Databases of PubMed,CNKI,ScienceDirect and Wanfang were searched,and the literatures were selected from 1990 to 2014,to review the studies of tolvaptan on autosomal dominant polycystic kidneydisease(ADPKD).ADPKD is a chronic progressive disease which significantly enhance the economy burden and death rate.No specific drug can be used to treat or prevent the progress of ADPKD.Tolvaptan applicated in early phase could prolong the progress of ADPKD,but with frequent and serious adverse events.【总页数】3页(P184-186)【作者】刘玉法;陈广新;刘纳新;苏立军【作者单位】山东省淄博市中心医院急诊科,山东淄博 255036;山东省淄博市中心医院急诊科,山东淄博 255036;山东省淄博市中心医院急诊科,山东淄博255036;山东省淄博市中心医院急诊科,山东淄博 255036【正文语种】中文【相关文献】1.中药三棱为主治疗常染色体显性遗传性多囊性肾病 [J], 李芳;贾丹兵;路更;李宇颖2.常染色体显性多囊肾病患者继发囊内严重感染一例 [J], 刘春莹;冯露夷;冷伟;任艳芸;谭颖颖;陈磊鑫;屈直;尚乘3.多囊蛋白-1在肾透明细胞癌及常染色体显性多囊肾病患者体液中含量的比较研究 [J], 吴俊;梅长林;赵海丹;戴兵;刘亚伟4.常染色体显性遗传性多囊性肾病发病机制的研究进展及治疗前景 [J], 刘海军;马云航5.西罗莫司无益于常染色体显性多囊性肾病的治疗 [J], Serra AL Poster D Kistler AD因版权原因,仅展示原文概要,查看原文内容请购买。