怎样分析气相色谱图
- 格式:doc
- 大小:14.00 KB
- 文档页数:2





气相色谱分析-定性分析方法气相色谱的定性分析就是要确定色谱图中每个色谱峰毕竟代表什么组分,因此必需了解每个色谱峰位置的表示办法及定性分析的办法。
(一)常用的保留值简介在气相色谱分析中,常用的保留值为保留时光tR、调节保留时光t'R、保留体积VR、调节保留体积V'R、相对保留值ris、比保留体积从和保留指数Ix。
各种保留值的计算公式如下: 1.保留时光tR 2.调节保留时光t'R t'R=tR-tM 死时光tM与被测组分的性质无关。
因此以保留时光与死时光的差值,即调节保留时光t'R,作为被测组分的定性指标,具有更本质的含义。
t'R反映了被测组分和固定相的热力学性质,所以用调节保留时光t'R比用保留时光tR作为定性指标要更好一些。
3.保留体积VR VR=tRFc 4.调节保留体积V'R V'R =(tR-tM)Fc=t'RFc=VR-VM 5.相对保留值ris 为了抵消色谱操作条件的变幻对保留值的影响,可将某一物质的调节保留时光:t'R(i)与一标准物(如正壬烷)的调节保留时光:t'R(s)相比,即为相对保留值(如相对壬烷值) 相对保留值ris仅与固定相的性质和柱温有关,与色谱分析的其它操作因素无关,因此具有通用性。
6.比保留体积Vg 比保留体积是气相色谱分析中的另一个重要保留值,其可按下式计算:式中t'R(i)—i组分的调节保留时光,min; m—固定液的质量,g;—在柱温、柱压下,柱内载气的平均体积流速; F'0—室温下由皂膜流量计测得的载气流速,ML/min; Tc—柱温,K; T0—室温,K; p0—室温下的大气压力,Pa; pw—室温下的饱和水蒸气压,pa; j—压力校正因子。
7.科瓦茨(Kovats)保留指数Ix 科瓦茨保留指数是气相色谱领域现已被广泛采纳的一定性指标,其规定为:在任一色谱分析操作条件下,对碳数为n的任何正构烷烃,其保留指数为100n。

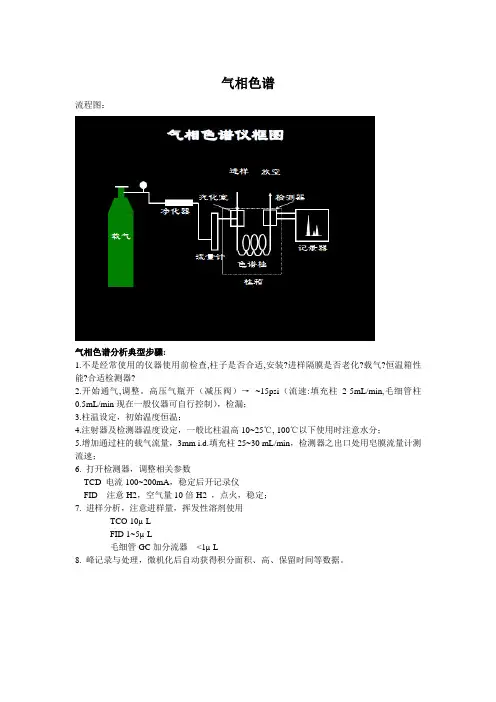
气相色谱
流程图:
气相色谱分析典型步骤:
1.不是经常使用的仪器使用前检查,柱子是否合适,安装?进样隔膜是否老化?载气?恒温箱性能?合适检测器?
2.开始通气,调整。
高压气瓶开(减压阀)→~15psi(流速:填充柱2-5mL/min,毛细管柱0.5mL/min现在一般仪器可自行控制),检漏;
3.柱温设定,初始温度恒温;
4.注射器及检测器温度设定,一般比柱温高10~25℃, 100℃以下使用时注意水分;
5.增加通过柱的载气流量,3mm i.d.填充柱25~30 mL/min,检测器之出口处用皂膜流量计测流速;
6. 打开检测器,调整相关参数
TCD 电流100~200mA,稳定后开记录仪
FID 注意H2,空气量10倍H2 ,点火,稳定;
7. 进样分析,注意进样量,挥发性溶剂使用
TCO 10µ L
FID 1~5µ L
毛细管GC加分流器<1µ L
8. 峰记录与处理,微机化后自动获得积分面积、高、保留时间等数据。



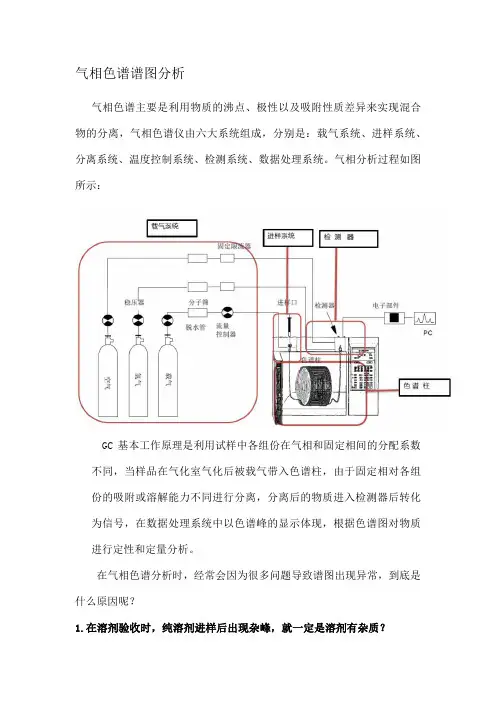
气相色谱谱图分析气相色谱主要是利用物质的沸点、极性以及吸附性质差异来实现混合物的分离,气相色谱仪由六大系统组成,分别是:载气系统、进样系统、分离系统、温度控制系统、检测系统、数据处理系统。
气相分析过程如图所示:GC基本工作原理是利用试样中各组份在气相和固定相间的分配系数不同,当样品在气化室气化后被载气带入色谱柱,由于固定相对各组份的吸附或溶解能力不同进行分离,分离后的物质进入检测器后转化为信号,在数据处理系统中以色谱峰的显示体现,根据色谱图对物质进行定性和定量分析。
在气相色谱分析时,经常会因为很多问题导致谱图出现异常,到底是什么原因呢?1.在溶剂验收时,纯溶剂进样后出现杂峰,就一定是溶剂有杂质?如果进空白针后也存在杂峰,连续进针后,峰面积逐渐减少,优先考虑仪器系统流路问题出现的杂峰。
可以从以下几个方面逐一排查:1)气源是否有问题;2)进样针,洗针瓶,隔垫,衬管,分流平板是否有污染;3)色谱柱是否有污染;4)检测器是否有污染等。
2.出现前沿峰1)样品过载,需稀释样品,减少进样量;2)载气流速过高;3)柱温太低,升高柱温;4)气化室温度太低;5)可能存在干扰峰,需要优化色谱条件;6)色谱柱选型错误,老化程度不够等。
3.出现拖尾峰1)衬管、分流平板或色谱柱被污染,或色谱柱安装不当,存在死体积;2)柱温或进样器温度低,升高温度;3)载气流量偏低;4)进样量大,减少进样量货增大分流比;5)进样器或气化室被高沸点杂质或残留污染等。
4.出现鬼峰1)色谱柱有残留,未完全老化;2)气化室、注射针等被污染或载气纯度不够;3)气化温度过高使样品某些组分分解;4)样品中有空气或TCD、ECD等密封性差(有漏气)等。
5.操作条件不变,原来可以分离的峰不见了?1)色谱柱被污染或者失效;2)载气系统被污染(载气纯度低或过滤器失效);3)注射垫或注射针漏气等。
6.进样后不出峰或者峰很小?1)检查检测器的信号值,信号值正常时,优先考虑进样口问题;2)进样针漏气或者堵塞;3)进样温度太低导致样品不能气化或柱温太低,导致样品在柱中冷凝;4)如果是FID,需要检查FID火焰是否点燃等。
聊一聊气相色谱仪N种图谱分析方法谱图分析(一)保留时间重现性差⊙指仪器工作条件和样品分析条件等均没有变化的情况下,保留时间变化较大、重现性较差。
A.色谱柱的一部分是否与柱箱内壁的金属面存在接触现象。
B.进样垫、色谱柱、过渡衬管的安装连接处是否存在漏气现象。
C.载气的输入压力是否正常。
D.载气流量是否正常或出现变化。
E.进样器、柱箱、检测器等的温度是否稳定。
F.如果保留时间与峰高/峰面积的重现性同时变差,则进行了上述检查后再参照[峰高/峰面积重现性差]中的各项进行检查。
注意:如果载气的流量、分流比、色谱柱温度等有变动时,保留时间或峰高/峰面积一定会起变化。
谱图分析(二)峰高/峰面积重现性差⊙指仪器工作条件和样品分析条件等均没有变化的情况下,峰高/峰面积变化较大、重现性较差。
A.注射器的性能是否正常以及进样时是否存在操作失误。
B.样品浓度(特别是挥发性样品)是否因放置时间过长而起变化。
C.各种气体的输入压力是否正常。
D.各种气体的流量是否正常或出现变化。
E.进样器、柱箱、检测器等的温度是否稳定。
F.如果峰高/峰面积与保留时间的重现性同时变差,在进行了上述检查后再参照[保留时间重现性差]中的各项进行检查注意:如果载气的流量、分流比、色谱柱温度等有变动时,保留时间或峰高/峰面积一定会起变化。
谱图分析(三)加出现刀锋⊙指样品出峰时上升缓慢而下降迅速,形如刀状。
A.减少样品的进样量。
B.提高色谱柱箱的温度。
C.改用较大内径的色谱柱。
D.增加固定液的涂层的厚度。
E.选用样品的溶解度较高的固定液。
F.尝试提高进样器的温度,改善峰的形状。
谱图分析(四)出钝峰⊙指所出的样品峰不尖,所有峰或一部分峰的顶部呈不规则形状(平头或园形)。
A.进样量太大使色谱柱或检测器形成饱和,减少进样量或降低样品浓度。
B.进样器是否存在漏气现象或玻璃衬管是否存在破损现象。
C.采用分流进样方式时,检查分流比及分析条件的设置是否正确。
D.采用不分流进样方式时,检查分析条件的设置是否正确。
怎样分析气相色谱图 The manuscript was revised on the evening of 2021在实际工作中,当我们拿到一个样品,我们该怎样定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤:1、样品的来源和预处理方法GC能直接分析的样品通常是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。
这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。
如果样品体系简单,试样组分可汽化则可直接分析。
如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,如采用吸附、解析、萃取、浓缩、稀释、提纯、衍生化等方法处理样品。
2、确定仪器配置所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。
一般应首先确定检测器类型。
碳氢化合物常选择FID检测器,含电负性基团(F、Cl等)较多且碳氢含量较少的物质易选择ECD检测器;对检测灵敏度要求不高,或含有非碳氢化合物组分时,可选择TCD检测器;对于含硫、磷的样品可选择FPD检测器。
对于液体样品可选择隔膜垫进样方式,气体样品可采用六通阀或吸附热解析进样方法,一般色谱仅配置隔膜垫进样方式,所以气体样品可采用吸附-溶剂解析-隔膜垫进样的方式进行分析。
根据待测组分性质选择适合的色谱柱,一般遵循相似相容规律。
分离非极性物质时选择非极性色谱柱,分离极性物质时选择极性色谱柱。
色谱柱确定后,根据样本中待测组分的分配系数的差值情况,确定色谱柱工作温度,简单体系采用等温方式,分配系数相差较大的复杂体系采用程序升温方式进行分析。
常用的载气有氢气、氮气、氦气等。
氢气、氦气的分子量较小常作为填充柱色谱的载气;氮气的分子量较大,常作为毛细管气相色谱的载气;气相色谱质谱用氦气作为载气。
3、确定初始操作条件当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。
在实际工作中,当我们拿到一个样品,我们该怎样定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤:
1、样品的来源和预处理方法
GC能直接分析的样品通常是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。
这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。
如果样品体系简单,试样组分可汽化则可直接分析。
如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,如采用吸附、解析、萃取、浓缩、稀释、提纯、衍生化等方法处理样品。
2、确定仪器配置
所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。
一般应首先确定检测器类型。
碳氢化合物常选择FID检测器,含电负性基团(F、Cl等)较多且碳氢含量较少的物质易选择ECD检测器;对检测灵敏度要求不高,或含有非碳氢化合物组分时,可选择TCD检测器;对于含硫、磷的样品可选择FPD检测器。
对于液体样品可选择隔膜垫进样方式,气体样品可采用六通阀或吸附热解析进样方法,一般色谱仅配置隔膜垫进样方式,所以气体样品可采用吸附-溶剂解析-隔膜垫进样的方式进行分析。
根据待测组分性质选择适合的色谱柱,一般遵循相似相容规律。
分离非极性物质时选择非极性色谱柱,分离极性物质时选择极性色谱柱。
色谱柱确定后,根据样本中待测组分的分配系数的差值情况,确定色谱柱工作温度,简单体系采用等温方式,分配系数相差较大的复杂体系采用程序升温方式进行分析。
常用的载气有氢气、氮气、氦气等。
氢气、氦气的分子量较小常作为填充柱色谱的载气;氮气的分子量较大,常作为毛细管气相色谱的载气;气相色谱质谱用氦气作为载气。
3、确定初始操作条件
当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。
这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。
进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。
样品浓度不超过10mg/mL时填充柱的进样量通常为1-5uL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2uL。
进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。
原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解温度。
4、分离条件优化
分离条件优化目的就是要在最短的分析时间内达到符合要求的分离结果。
在改变柱温和载气流速也达不到基线分离的目的时,就应更换更长的色谱柱,甚至更换不同固定相的色谱柱,因为在GC中,色谱柱是分离成败的关键。
5、定性鉴定
所谓定性鉴定就是确定色谱峰的归属。
对于简单的样品,可通过标准物质对照来定性。
就是在相同的色谱条件下,分别注射标准样品和实际样品,根据保留值即可确定色谱图上哪个峰是要分析的组分。
定性时必须注意,在同一色谱柱上,不同化合物可能有相同的保留值,所以,对未知样品的定性仅仅用一个保留数据是不够的,双柱或多柱保留指数定性是GC中较为可靠的方法,因为不同的化合物在不同的色谱柱上具有相同保留值的几率要小得多。
条件允许时可采用气相色谱质谱联机定性。
6、定量分析
要确定用什么定量方法来测定待测组分的含量。
常用的色谱定量方法不外乎峰面积(峰高)百分比法、归一化法、内标法、外标法和标准加入法(又叫叠加法)。
峰面积(峰高)百分比法最简单,但最不准确。
只有样品由同系物组成、或者只是为了粗略地定量时该法才是可选择的。
相比而言,内标法的定量精度最高,因为它是用相对于标准物(叫内标物)的响应值来定量的,而内标物要分别加到标准样品和未知样品中,这样就可抵消由于操作条件(包括进样量)的波动带来的误差。
至于标准加入法,是在未知样品中定量加入待测物的标准品,然后根据峰面积(或峰高)的增加量来进行定量计算。
其样品制备过程与内标法类似但计算原理则完全是来自外标法。
标准加入法定量精度应该介于内标法和外标法之间。
7、方法的验证
所谓的方法验证,就是要证明所开发方法的实用性和可靠性。
实用性一般指所用仪器配置是否全部可作为商品购得,样品处理方法是否简单易操作,分析时间是否合理,分析成本是否可被同行接受等。
可靠性则包括定量的线性范围、检测限、方法回收率、重复性、重现性和准确度等。