
Universal Kinase Activity Kit
Catalog Number: EA004 The protocol presented is for a 96-well format. Materials provided are sufficient for two 96-well microplates or equivalent. This package insert must be read in its entirety before using this product.
ASSAY PRINCIPLE
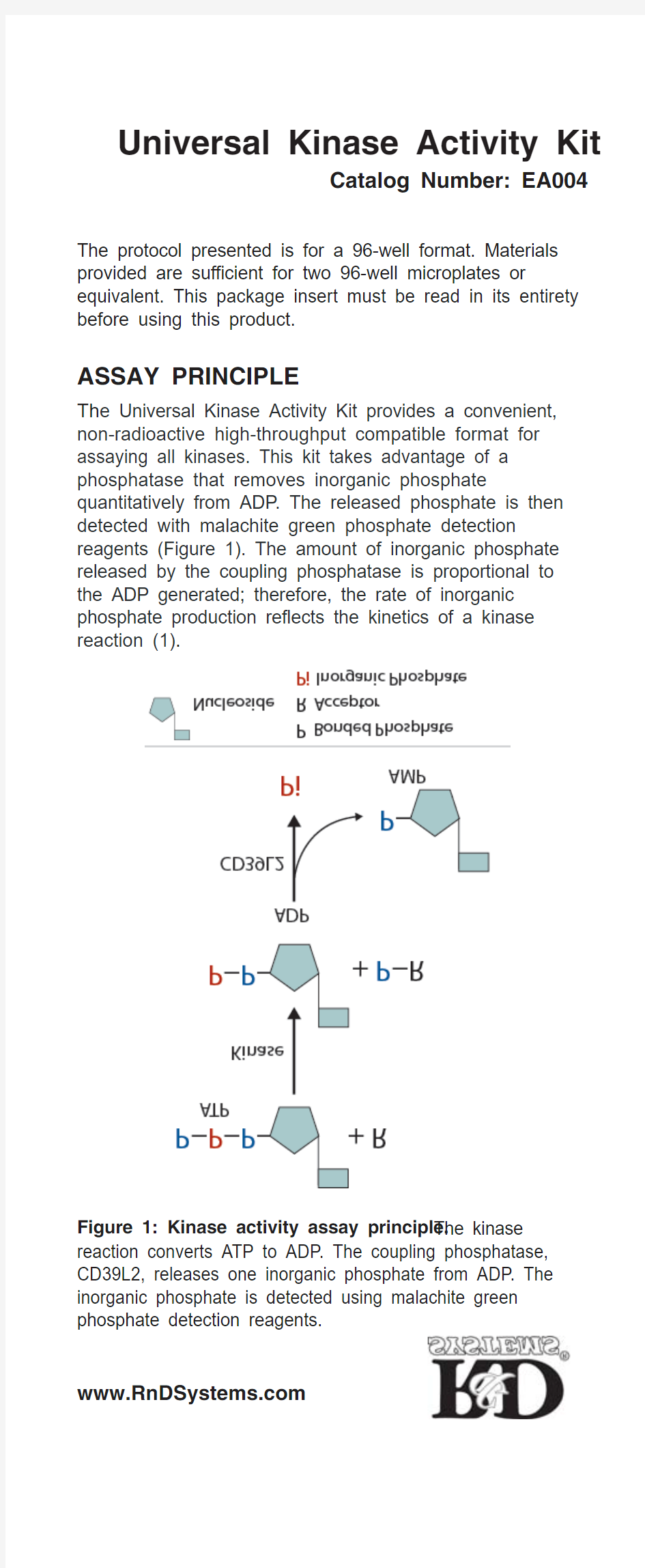
Th e Universal Kinase Activity Kit provides a convenient, non-radioactive high-throughput compatible format for assaying all kinases. This kit takes advantage of a phosphatase that removes inorganic phosphate quantitatively from ADP. The released phosphate is then detected with malachite green phosphate detection reagents (Figure 1). The amount of inorganic phosphate released by the coupling phosphatase is proportional to the ADP generated; therefore, the rate of inorganic phosphate production reflects the kinetics of a kinase reaction (1).
Figure 1: Kinase activity assay principle.The kinase reaction converts ATP to ADP. The coupling phosphatase, CD39L2, releases one inorganic phosphate from ADP. The inorganic phosphate is detected using malachite green phosphate detection reagents.
https://www.doczj.com/doc/ac14428438.html,
MATERIALS PROVIDED
Upon receipt, store ATP, ADP, and Coupling Phosphatase 4 at£-20 °C until use. All other components may be stored at ambient temperature. Use before the kit expiration date.
Coupling Phosphatase 4(Part 895988, 1 vial)- CHO cell-expressed recombinant mouse CD39L2/ENTPD6; 200m L of 100 ng/m L in 25 mM Tris, 150 mM NaCl, 5 mM CaCl2, 20% glycerol, pH 7.5.
Phosphatase Buffer 4(Part 895989, 2 vials)- Each vial contains 1.5 mL of a 10X solution of 250 mM HEPES, 1.5 M NaCl, 100 mM MgCl2, 100 mM CaCl2, pH 7.0.
ATP(Part 895990, 2 vials)- Each vial contains 200L of 10 mM ATP in 25 mM sodium borate, pH 9.0.
ADP(Part 895991, 1 vial)- 200m L of 10 mM ADP in
25 mM sodium borate, pH 9.0.
Phosphate Standard(Part 892809, 1 vial)- 200m L of
1 mM phosphate (KH2PO4) in deionized water.
Malachite Green Reagent A(Part 895855, 3 vials)-Each vial contains 3 mL of ammonium molybdate in 3 M sulfuric acid.
Malachite Green Reagent B(Part 895856, 3 vials)-Each vial contains 3 mL of malachite green oxalate and polyvinyl alcohol.
MATERIALS REQUIRED
·Kinase.
·Acceptor substrate for kinase.
·Assay buffer (if different than provided).·Deionized or distilled water.
·Microplate reader capable of measuring absorbance at 620 nm.
·Microplate (R&D Systems, Catalog # DY990 or equivalent).
PRECAUTION
Malachite Green Reagent A, Malachite Green Reagent B, and Phosphate Standard supplied with this kit are acidic solutions. Wear protective gloves, clothing, eye, and face protection. Wash hands thoroughly after handling.
REAGENT PREPARATION
The volumes of working solutions below are for 20 kinase reactions. Adjust the volumes accordingly if a different number of reactions is planned. Discard the leftovers of the preparations after the assay is complete.
1X Assay Buffer- Add 200m L of 10X Phosphatase Assay Buffer 4 to 1.8 mL of deionized or distilled water in a tube. Mix well.
Prepare the following items in the 1X Assay Buffer.·Kinase: 300m L at 1 ng/m L to 1m g/m L
·Acceptor Substrate: 300m L at 0.5-5 mM
·ADP: 100L at 1 mM
·ATP: 100L at 1 mM
·Coupling Phosphatase 4: 200m L at 10 ng/L
PHOSPHATE STANDARD CURVE DETERMINATION
This protocol is for determining a standard curve in a
96-well microplate. It is recommended that the standards be assayed in duplicate and a standard curve be generated before an enzyme kinetic assay.
1.Add 40m L of the 1 mM Phosphate Standard to 360m L
of 1X Assay Buffer in a microcentrifuge tube and mix well. Transfer 200m L of the dilution into 200m L of
1X Assay Buffer in a second tube. Repeat the process to prepare a 2-fold serial dilution. The eighth tube
contains only 1X Assay Buffer and serves as the
blank.
2.Transfer 50L of each dilution into microplate wells in
duplicate.
3.Add 30m L of Malachite Green Reagent A to each
well. Mix by gently tapping the plate.
4.Add 100m L of deionized or distilled water to each
well.
5.Add 30m L of Malachite Green Reagent B to each
well. Mix by gently tapping the plate.
6.Incubate the plate for 20 minutes at room temperature
to stabilize the color development. The yellow
background will fade during incubation.
7.Determine the optical density (OD) of each well using
a microplate reader set to 620 nm.
8.Average the duplicate readings for each standard and
subtract the OD of the average zero standard. Plot
phosphate input (pmol) vs. the corrected OD
(Figure 2).
Figure 2:A phosphate standard curve determined using 1X Assay Buffer.The slope of the linear regression line, 3504.3 pmol/OD, represents the amount of phosphate corresponding to a unit of absorbance at 620 nm. It is referred to as the phosphate conversion factor (CF) in subsequent calculations. CF may vary under different conditions. This standard curve is provided for demonstration only.
KINASE ASSAY PROTOCOL
This standard kinase assay is carried out in 50m L for
10 minutes at room temperature and is coupled to 100 ng of Coupling Phosphatase 4. The reaction may be scaled up proportionally. 1X Assay Buffer can be used in most kinase reactions. Individual kinase reactions can be optimized by adjusting the concentrations of kinase, substrates, and metal ions. It is recommended that all samples be assayed in duplicate. If assay conditions change, the amount of coupling enzyme should be adjusted to reach the optimal coupling rate. For more information, see the Technical Hints and Limitations section.
1.Prepare the substrate mix using the freshly prepared
working solutions.
ATP10m L
Acceptor15m L
Total Volume25m L
2.Prepare the enzyme mix using the freshly prepared
working solutions.
Coupling Phosphatase 410m L
Kinase15m L
Total Volume25m L
3.For a positive control, use ADP in place of ATP.
4.For a negative control, use 1X Assay Buffer in place of
the kinase.
5.Initiate the reaction by adding the substrate mix to the
enzyme mix in a 96-well microplate.
6.As an assay blank, add 50m L of 1X Assay Buffer in a
separate well.
7.Incubate the microplate for 10 minutes at room
temperature.
8.Terminate the reactions by adding 30L* of Malachite
Green Reagent A to each well. Mix by gently tapping the microplate.
9.Add 100L* of deionized or distilled water to each
well. Mix gently by tapping the microplate.
10.Add 30L* of Malachite Green Reagent B to each
well. Mix gently by tapping the microplate.
11.Incubate the microplate for 20 minutes at room
temperature to stabilize the color development.
12.Determine the optical density of each well using a
microplate reader set to 620 nm, and adjust the OD by subtracting the negative control.
13.Calculate the phosphate in each reaction using the
previously determined conversion factor.
*If altering the reaction volume, it is necessary to maintain a ratio of the reaction volume plus water : Malachite Green Reagent A : Malachite Green Reagent B of 5:1:1.
ASSAY EXAMPLE
Figure 3: PKA C b assay.Recombinant human PKA C b (R&D Systems, Catalog # 4596-KS) was assayed with 0.2 mM ATP, 0.4 mM Kemptide (https://www.doczj.com/doc/ac14428438.html,, Catalog # 22595), and 0.1m g of Coupling Phosphatase 4 in 50m L of 1X Assay Buffer for 10 minutes at room temperature. When the corrected optical density was plotted versus the amount of kinase, a slope of
0.908 OD/m g was obtained. Using the CF of
3504 pmol/OD determined in Figure 2 and the equation (specific activity = slope x (CF/time)), the specific activity was calculated to be 318 pmol/min/m g. After correction with the coupling rate, 0.475 (Table 1), the activity is
670 pmol/min/m g.
TECHNICAL HINTS AND LIMITATIONS ·Malachite Green Reagents are highly sensitive to phosphate. All reagents must be phosphate-free. In
particular,phosphate-containing buffers should be
avoided at all times.If a phosphate-containing enzyme preparation is to be assayed, its phosphate content
should be removed using dialysis or chromatography
step s.
·The linear response region for phosphate detection is between 100-4000 pmol. Higher levels of phosphate may cause precipitation of the phosphate-malachite complex.
To ensure that the phosphate content falls into this range,
a portion of the reactions may be used for detection.
Alternatively, the reactions can be diluted prior to
detection. In any case, the ratio of reaction plus water :
Malachite Green Reagent A : Malachite Green Reagent B should be kept at 5:1:1.
·Although CD39L2 prefers ADP as a substrate, it has slight activity on ATP. Increasing the amount of coupling phosphatase will increase the hydrolysis of ATP,
therefore resulting in higher background.To reduce the background while maintaining a good signal, the
amount of coupling phosphatase needs to be
restricted to achieve an optimal coupling rate
between 0.4 and 0.6.A coupling rate is defined as the
ratio of the product ADP that has been converted to the
signal of free inorganic phosphate (1). The coupling rate of a kinase reaction is used to calculate the amount of
ADP produced by the kinase reaction based on the
amount of phosphate detected. Coupling rates for assays under different ATP concentrations using the provided
protocol are listed in Table 1.
·The coupling rate is pH, NaCl, ATP, and temperature sensitive and Ca2+dependent (1). If these conditions are changed, the coupling rate should be recalculated, which further requires the determination of the rate constant of
the coupling enzyme. For rate constant determination and coupling rate calculation, see reference 1 or the technical information available on https://www.doczj.com/doc/ac14428438.html,.·ATP is less stable under the acidic conditions of Malachite Green Reagents, which will result in slowly
increasing background with time. For consistency, all
reactions should be read 20 minutes after the addition of Malachite Green Reagents.
·If the reaction conditions for a kinase and the coupling phosphatase are not compatible, a decoupled assay is
suggested, in which phosphatase and phosphatase
buffer are added into the kinase reaction for an additional 10-20 minutes. In this case, the strength of the
phosphatase buffer is recommended to be 4X higher than that of the kinase buffer.
· A decoupled assay is also recommended for kinases that have low activities or require reaction time of more than
30 minutes to avoid high background from ATP hydrolysis
by CD39L2.
·DTT may be required for a protein kinase assay and can be added into a kinase reaction at 1 mM without adverse effect to the coupling enzyme and the phosphate
detection.
·Pipetting concentrated proteins or polypeptides can cause foaming, which should be eliminated before
measurement.
Table 1: Coupling rates for 10 minute 50m L reactions using the supplied buffer at room temperature. Coupling rates under different ATP concentations and varying amounts of CD39L2 are listed. The coupling rate is a function of the rate constant, reaction volume, and reaction time. Calculations are based on the specific rate constant of 97.78 nmol/min/m g/mM. For best performance, coupling rates are suggested to be between 0.4 and 0.6. For calculating coupling rates under different assay conditions, such as incubation time or the amount of coupling enzyme, refer to reference 1 or technical information available at https://www.doczj.com/doc/ac14428438.html,.
REFERENCE
1.Wu, Z.L. (2011) PLoS ONE6(8):e2317
2.
APPENDIX
The following detergents and common reagents were tested for interference in the phosphate assay. The effects occurred at concentrations above those listed.
Detergents1Level Effect
TritonòX-1000.3%Increased Blank
Tweenò200.1%Reduced Sensitivity NP-40 Alternative1%None
CHAPS1%None
SDS0.01%Increased Blank Deoxycholate0.01%Increased Blank
Precipitates Common Reagents1Level Effect
Glycerol5%Reduced Sensitivity
DMSO10%Reduced Sensitivity
Ethanol25%Reduced Sensitivity
BSA0.03 mg/mL Reduced Sensitivity
EDTA10 mM None Dithiothreitol 3 mM Reduced Sensitivity b-mercaptoethanol10 mM None
Na3VO4 1 mM Reduced Sensitivity
NaF10 mM None
NaCl100 mM None
KCl100 mM None
CaCl210 mM None
1Tested using the microplate assay protocol in 25 mM Tris-HCl, pH 7.5, with or without 1 nmol phosphate (KH2PO4).
All trademarks and registered trademarks are the property of their respective owners.
FOR RESEARCH USE ONLY.
NOT FOR USE IN DIAGNOSTIC PROCEDURES.
USA & Canada|R&D Systems, Inc.
614 McKinley Place NE, Minneapolis, MN 55413, USA
TEL: (800) 343-7475 (612) 379-2956 FAX: (612) 656-4400
UK & Europe|R&D Systems Europe, Ltd.
19 Barton Lane, Abingdon Science Park, Abingdon OX14 3NB, UK TEL: +44 (0)1235 529449 FAX: +44 (0)1235 533420
China|R&D Systems China Co., Ltd.
24A1 Hua Min Empire Plaza, 726 West Yan An Road
Shanghai PRC 200050
TEL: +86 (21) 52380373 FAX: +86 (21) 52371001
726180.04/12
SOD(超氧化物歧化酶)活性测定 氮蓝四唑法 一、原理 超氧化物歧化酶(superoxide dismutase ,SOD)普遍存在动、植物的体内,是一种清除超氧阴离子自由基的酶,它催化下面的反应: o 2.-+H O 2 22+O H + 反应产物H 2O 2可由过氧化氢酶进一步分解或被过氧化物酶利用。超氧化物歧化酶抑制氮蓝四唑(NBT)在光下的还原作用来确定酶活性的大小。在有氧化物质存在下,核黄素可被光还原,被还原的核黄素在有氧条件下极易被氧化而产生超氧阴离子,超氧阴离子可将氮蓝四唑还原为蓝色的甲腙,后者在560nm 处有最大吸收。而SOD 可清除超氧阴离子,从而抑制了甲腙的形成。于是光还原反应后,反应液蓝色愈深,说明酶的活性愈低,反之酶的活性俞高。据此可计算出酶活性的大小。 二、材料、仪器设备及试剂 (一)材料 植物器官(花瓣、叶片等) (二)仪器设备 冰箱、低温高速离心机、微量加样器 (1mL 、20μL 、100μL)、移液管、精密电子天平、UV-752型紫外分光光度计、试管、研钵、剪刀、镊子、荧光灯(反应试管处照度为4000Lux 或Lx) (三)试剂 (1) 0.05mol/L 磷酸缓冲液(PH7.8)。 (2) 130mmol/L 甲硫氨酸(Met)溶液:称1.9399gMet 用磷酸缓冲液定溶至100mL 。 (3)750μmol/L 氮蓝四唑溶液:称取0.06133gNBT 用磷酸缓冲液定溶至100mL ,避光保存。 (4)100μmol/LEDTA -Na 2溶液:称取0.03721g EDTA-Na 2,用磷酸缓冲液定溶1000mL 。 (5)20μmol/L 核黄素溶液:称取0.0753g 核黄素用蒸馏水定溶到1000mL ,避光保存。 三、试验步骤 (一)酶液的提取 (1)称取植物材料(去叶脉)0.2g ,加1ml 预冷的磷酸缓冲液在冰浴上研磨成浆,加缓冲
细胞增殖及细胞活力检测方法 目前主要有两种用于检测细胞增殖能力的方法。一种是直接的方法,通过直接测定进行分裂的细胞数来评价细胞的增殖能力。另一种是间接的方法,即细胞活力(cell viability)检测方法,通过检测样品中健康细胞的数目来评价细胞的增殖能力。显然,细胞活力检测法并不能最终证明检测样品中的细胞是否在增殖。如细胞在某一培养条件下会自发启动凋亡程序,但药物的干扰可抑制凋亡的发生;这时若采用细胞活力检测法,显然可以区分两种条件下的细胞数量,但我们并不能从药物干扰组细胞数大于对照组的事实说明药物可促进细胞增殖的结论。所以最直接的证据应该采用方法一。 用于检测细胞增殖能力最经典的方法是用氚标记的胸腺嘧啶核苷处理细胞,再检测DNA链中氚含量。若细胞具有增殖能力,DNA合成过程中将会采用氚标记的胸腺嘧啶核苷作为合成原料,因此检测细胞DNA链内标记核苷酸的量可判断细胞是否进行DNA 的合成。 但更为常用的方法是BrdU检测法。用BrdU预处理的细胞中,BrdU可代替胸腺嘧啶核苷插入复制的DNA双链中,而且这种置换可以稳定存在,并带到子代细胞中。细胞经过固定和变性处理后,可用免疫学方法检测DNA中BrdU的含量(如采用鼠抗BrdU单克隆抗体特异识别BrdU,再采用辣根过氧化酶标记的山羊抗鼠IgG二抗标记,最后用比色法或荧光的方法进行定量测定),从而判断细胞的增殖能力。Calbiochem/EMD公司提供一种BrdU检测试剂盒,以微孔板的形式,合并所有清洗、固定、变性的步骤以单一试剂当中。比色检测在一抗二抗标记后在450nm下读数,
所有操作在3小时内结束。而且该试剂盒的灵敏度与市场上其他同类产品相比是最强的。1000个细胞以上水平的检测只需用BrdU预孵育2小时,100个细胞则采用过夜预孵育,即可检测细胞的增殖能力。 BrdU法的一个缺点是需要固定和变性等破坏DNA的处理。有些情况下,研究者可能希望在测定细胞增殖能力的同时检测细胞的总DNA含量,然而,在变性条件下,DNA 的双链结构将被破坏,DAPI和Hoechest 33342等核酸标记探针就不再能识别DNA,因而也无法估计DNA总量。Molecular Probes公司的Click-iT EdU检测试剂盒可以解决这个问题。这种方法不需要变性步骤,因为荧光探针标记的叠氮化物小分子,而不是庞大的抗体分子,可以很轻易的识别并结合未变性DNA双链中的EdU分子。采用BrdU方法时,你必须非常小心的去对DNA进行变性,才能一方面使BrdU抗体进入细胞,另一方面又保留足够的双链DNA分子来进行细胞周期的分析。有了EdU 后则不同,由于你不再需要变性,这一切都很简单了;另外,常常用于DNA变性的HCl,可能破细胞内坏蛋白的抗原识别位点,因而限制了BrdU检测法中同时检测其他蛋白的应用,但这种情况在EdU法中不存在。 图1:EdU及BrdU原理示意图(摘至invitrogen说明书) 在一些情况下,细胞活力的检测相当于细胞增殖能力的测定。用于细胞活力检测的方法又很多,这些方法主要采用特殊的试剂来测定细胞的代谢活力,Alamar Blue,MTT及其他四唑盐。它们通过检测细胞的氧化还原活性来检测细胞增殖能力,所以这是一种间接的方法。 Calbiochem的快速细胞增殖试剂盒,或者,严格来说,叫细胞活力试剂盒,采用一
连苯三酚自氧化法测定·O2-自由基的清除能力简介(适用于:SOD及各种抗氧化剂) 文献来源 [1] Xican Li. Improved Pyrogallol Autoxidation Method: A Reliable and Cheap Superoxide-scavenging Assay Suitable for All Antioxidants. Journal of Agricultural and Food Chemistry, 2012, 60:6418-6424. 操作图解 具体方法 1 溶液配制 1.1 Tris溶液(0.1mol/L):1.21 gTris(三羟甲基氨基甲烷,M.W. 121.1)+100 mL蒸馏水。 1.2 HCl溶液(0.1mol/L):取0.1 mL浓盐酸,加蒸馏水稀释到6 mL。 1.3 Tris-HCl缓冲液(0.05mol/L,pH7.4,含1mmol/L Na2EDTA) 40 mL0.1 mol/L Tris溶液+ x mL0.1 mol/L HCl溶液+15.2 mg Na2EDTA,混合,稀释到80 mL。用pH 计测量,pH应为7.4。用棕色瓶保存在冰箱内(最多保存三天) 。(以上为一个样品的用量)用前稍热至室温,再测pH值,符合要求即可。 1.4 60 mmol/L连苯三酚溶液(溶于1 mmol/L盐酸中) 取0.1mol/L HCl溶液(见1.2项)20μL,用蒸馏水稀释到2 mL,得1 mmol/L盐酸溶液(用pH计测量,pH=2.5-3.0)。再往里加连苯三酚14.6 mg (M。W.126.1 ),即得。(当天有效,以上为1个样品的用量)。 2 测试液 2.1连苯三酚溶液:取2950μL Tris-HCl缓冲液加入到石英比色皿中,再加约50μL连苯三酚溶液,迅速混合(颠覆式),开始计时,每隔30秒读数一次A值(325nm),至300秒(5min)时为止。(空白参比:Tris-HCl 缓冲液) ΔA=A325nm,300s - A325nm,30s。由于ΔA值反映了生成·O2的初始浓度,所以,对于同一批实验而言,此时的ΔA值必须相等。此时的ΔA为ΔA0。 3.2 样品溶液:取xμL样品溶液加入到大石英比色皿中,再加(2950-x)μL Tris-HCl缓冲液,再加50μL 连苯三酚溶液,迅速混合(颠覆式),开始计时,每隔30秒读数一次(A值,325nm),至300秒时为止。(空白参比:Tris-HCl缓冲液) ΔA=A325nm,300s - A325nm,30s。此时的ΔA为ΔA样。 3 计算公式
酶液提取:称取鲜叶样品。0.5g于预冷的研钵中,加lml0.05mol/1 pH7.0磷酸缓冲液在冰浴上研磨成浆,加缓冲液使终体积为5ml。将提取液于10000转/分冷冻离心20分钟,上清液用于测定SOD, POD, CAT活力测定及丙二醛含量测定。 SOD: 测定SOD活性的试剂配制及用量: ①磷酸缓冲液(PH7.8) 0.05moI/L 3.1m1,空白为3.2m1; ②EDTA-Na 2 1mg/m1 0.2mL; ③L一甲硫氨酸20mg/mL 0.2m1; ④核黄素0.1 mg/ml,吸取上清液0.2m1; ⑤NBT lmg/ml 0.2m1; ⑥提取酶液0.1ml,空白不加酶液。 反应总体积为4m1, 第1组、4000Lx光照30min,遮黑布终止反应(用光照培养箱,灯管全部打开即可)。 第2组、黑暗处理30 min。 置560nm处测定光密度。SOD活性单位以抑制NBT光化还原50%作为一个酶活性单位(u),按以下公式计算SOD活性: 式中,Ack为照光管的吸光度值;AE为遮光管的吸光度值:V为样品液总体积((ml);Vt为测定时样品用量(ml); W为样品鲜重。 POD: 在试管中依次加入 4m1 0.3%愈创木酚(0.02mo1/1 pH6磷酸缓冲液配置)、 50ul酶液、 50 ul0.3%H 20 2, 摇匀,立即计时,1分钟后在470nm波长下比色,每1分钟记录一次吸光度值,连续记录5分钟。以每分钟内A470变化0.01为1个过氧化物酶活性单位(U),按下式计算过氧化物酶活性:
式中:d A470为反应时间内吸光度的变化:Vt为提取液总体积(ml); W为样品鲜重(g); Vs为测定时取用酶液体积(ml); t为反应时间(min) 。 CAT 取酶提取液50 u 1, 加入3m1 0.05 mol/1 pH7.0磷酸缓冲液, 再加入0.3%H 2O 2 200 u1, 迅速摇匀,立即计时,1分钟后在UV-754分光光度计的240nm波长下比色,每1分钟记录一次吸光度值,连续记录5分钟。以每分钟内A240下降0.01为1个酶活性单位(U),按下式计算过氧化氢酶活性: 式中:△A 240 为反应时间内吸光度的变化;Vt为提取液总体积(ml):w为样品鲜重(g); Vs为测定时取用酶液体积(ml); t为反应时间(min) MDA 取上清液1.5m1, 加入2.5m10.5%的硫代巴比妥酸(TBA)(用10%三氯乙酸配制)。 混合物于100℃沸水浴中加热20min,迅速冷却,于10000转/分离心20分钟,分别测定上清液在450, 532nm及600nm处的吸光度值。按以下公式计MDA浓度C( u mol/1)和含量(nmollgFW): C(u mol/1)=6.45 X (A532-A600)-0.56 X A450
抗氧化酶 S O D P O D C A T活性测 定方法 集团文件版本号:(M928-T898-M248-WU2669-I2896-DQ586-M1988)
抗氧化酶(SOD、POD、CAT)活性测定方法 酶液制备:取0.2g(可视情况调整)样品(新鲜叶片或根系)洗净后置于预冷的研钵中,加入1.6ml 50mmol/L预冷的磷酸缓冲液 (pH7.8)在冰浴上研磨成匀浆,转入离心管中在4℃、12000g下离心20min,上清液即为酶液。 一、超氧化物歧化酶(SOD)活性测定(氮蓝四唑光化还原法) 1、试剂的配制 (1)0.05mol/L磷酸缓冲液(PBS,pH7.8): A母液:0.2mol/L磷酸氢二钠溶液: 取Na 2HPO 4 ·12H 2 O(分子量 358.14)71.7g; B母液:0.2mol/L磷酸二氢钠溶液:取NaH 2PO 4 ·2H 2 O(分子量 156.01)31.2g。 分别用蒸馏水定容到1000ml。 0.05mol/L PBS(pH7.8)的配制:分别取A母液(Na 2HPO 4 ) 228.75ml,B母液(NaH 2PO 4 ) 21.25ml,用蒸馏水定容至1000ml。 参考文献:李合生主编:植物生理生化实验原理和技术.高等教育出版社,2000:267~268。 (2)130mM甲硫氨酸溶液:取1.9399g Met用磷酸缓冲液(pH7.8)定容至100ml。 (3)100μM EDTA-Na 2溶液:取0.03721gEDTA-Na 2 用磷酸缓冲液定 容至1000ml。 (4)20μM核黄素溶液:取0.0753g核黄素用蒸馏水液定容至1000ml,避光保存。 (5)750uM 氮蓝四唑(NBT)溶液:取0.06133g NBT用PBS定容至100ml,避光保存。 2、酶活性测定 (1)取10ml试管(要求透明度好)
细胞增殖及细胞活力检 测方法 Document number:NOCG-YUNOO-BUYTT-UU986-1986UT
细胞增殖及细胞活力检测方法 目前主要有两种用于检测细胞增殖能力的方法。一种是直接的方法,通过直接测定进行分裂的细胞数来评价细胞的增殖能力。另一种是间接的方法,即细胞活力(cell viability)检测方法,通过检测样品中健康细胞的数目来评价细胞的增殖能力。显然,细胞活力检测法并不能最终证明检测样品中的细胞是否在增殖。如细胞在某一培养条件下会自发启动凋亡程序,但药物的干扰可抑制凋亡的发生;这时若采用细胞活力检测法,显然可以区分两种条件下的细胞数量,但我们并不能从药物干扰组细胞数大于对照组的事实说明药物可促进细胞增殖的结论。所以最直接的证据应该采用方法一。 用于检测细胞增殖能力最经典的方法是用氚标记的胸腺嘧啶核苷处理细胞,再检测DNA 链中氚含量。若细胞具有增殖能力,DNA合成过程中将会采用氚标记的胸腺嘧啶核苷作为合成原料,因此检测细胞DNA链内标记核苷酸的量可判断细胞是否进行DNA的合成。 但更为常用的方法是BrdU检测法。用BrdU预处理的细胞中,BrdU可代替胸腺嘧啶核苷插入复制的DNA双链中,而且这种置换可以稳定存在,并带到子代细胞中。细胞经过固定和变性处理后,可用免疫学方法检测DNA中BrdU的含量(如采用鼠抗BrdU单克隆抗体特异识别BrdU,再采用辣根过氧化酶标记的山羊抗鼠IgG二抗标记,最后用比色法或荧光的方法进行定量测定),从而判断细胞的增殖能力。 Calbiochem/EMD公司提供一种BrdU检测试剂盒,以微孔板的形式,合并所有清洗、固定、变性的步骤以单一试剂当中。比色检测在一抗二抗标记后在450nm下读数,所有操作在3小时内结束。而且该试剂盒的灵敏度与市场上其他同类产品相比是最强的。1000个细胞以上水平的检测只需用BrdU预孵育2小时,100个细胞则采用过夜预孵育,即可检测细胞的增殖能力。 BrdU法的一个缺点是需要固定和变性等破坏DNA的处理。有些情况下,研究者可能希望在测定细胞增殖能力的同时检测细胞的总DNA含量,然而,在变性条件下,DNA的双链结构将被破坏,DAPI和Hoechest 33342等核酸标记探针就不再能识别DNA,因而也无法估计DNA总量。Molecular Probes公司的Click-iT EdU检测试剂盒可以解决这个问题。这种方法不需要变性步骤,因为荧光探针标记的叠氮化物小分子,而不是庞大的抗体分子,可以很轻易的识别并结合未变性DNA双链中的EdU分子。 采用BrdU方法时,你必须非常小心的去对DNA进行变性,才能一方面使BrdU抗体进入细胞,另一方面又保留足够的双链DNA分子来进行细胞周期的分析。有了EdU后则不同,由于你不再需要变性,这一切都很简单了;另外,常常用于DNA变性的HCl,可能破细胞内坏蛋白的抗原识别位点,因而限制了BrdU检测法中同时检测其他蛋白的应用,但这种情况在EdU法中不存在。
SOD酶活性测定 所需药品: (1)0.1mol/l pH7.8的磷酸钠缓冲液: A液:0.1mol/l磷酸氢二钠液 B液:0.1mol/l磷酸二氢钠液 1毫升B+10.76毫升A (2)0.026mol/l蛋氨酸液(Met):现用现配 称取0.3879克蛋氨酸,用1号液定容至100毫升。 (3)75*10-5mol/l氯化硝基四氮唑蓝(NBT)液:现用现配 称取0.1533克NBT,先用少量蒸馏水溶解,然后定容至250毫升。 (4)1umol/lEDTA-2钠和2*10-5mol/l核黄素混合液 (5)0.05mol/l pH7.8的磷酸钠缓冲液 (6)石英砂 实验步骤: 1.酶液制备:称取0.5克鲜叶,放入研钵中,加入3毫升5号液和少量石英砂,于冰浴中研成匀浆。然后用5号液定容至8毫升,于0~4℃、13000g时离心15分钟,上清液即为酶提取液。酶液可在低于0℃下的环境中保存。 2.按下表加入试剂: 试剂摇匀后,迅速遮光处理1号杯,其余杯在25℃、光强为4000勒克司的条件下照光处理15分钟,然后立即遮光。接着在560nm下,以1号杯作为空白测定其余杯中溶液的光密度。假定2、3号杯中溶液抑制NBT光还原的相对百分率为100%,然后按下式分别计算其余杯中溶液抑制NBT光还原的相对百分率。 M/N=100/X M——2、3号杯中溶液的光密度的平均值 N——其余杯中溶液的光密度值 X——其余杯中溶液抑制NBT光还原的相对百分率 然后以酶液量为横坐标,以其余杯中溶液抑制NBT光还原的相对百分率(X)为纵坐标制作曲线,根据线性好的曲线所得出的函数关系计算抑制NBT光还原的相对百分率为50%时所加入的酶液量,以该酶液量作为1个酶活单位。 结果计算:SOD活力按下式计算: A=V*1000*60/(B*W*T)
细胞活性测定方法 细胞活性指标通常包括细胞膜对核酸染料的通透性,代谢活性,膜电位等。核酸染料有多种,如EB带有单个自由正电荷,能通过完整细胞膜。而PI,TO—PRO—1,TO-PRO-3等等带一个有四铵基团和两个或两个以上正电荷的染料是不能通过完整细胞膜进入细胞内的。因而吸收了这些多电荷染料的细胞被认为是非活性的。另外,一些酸性染料,如上面提到的台盼兰,曙红等都是膜非通透性。 代谢活性是另一重要指标,它通过细胞内的酶的活性来判定。使用细胞某种酶的底物,它能通过(或是不能通过)完整细胞膜,在细胞内被酶切而产生荧光性,膜不通透性产物,能在细胞膜完好的细胞内存留,在细胞膜不完整的细胞内散失很快。通过检测荧光强度就可知细胞代谢活性。FDA(fluorescein diacetate)和CTC(5-cyano-2,3-ditolyl tetrazolium chloride)是常用的两种底物。前者虽然透过细胞膜的速度较慢,但它的产物基本不往外通透。后者经细胞内脱氢酶催化而具有荧光性,能提供细胞呼吸代谢系统活性和细胞膜完整性信息。 正常细胞的细胞膜两侧维持着一个胞内为负的膜电位为梯度,带正电的亲脂性染料,如Cyanines类能因电梯度而通过细胞的脂双层膜聚积在活细胞内,带负电的亲脂性染料如oxonols会被排除在外。不再维持着膜电位梯度的细胞里,则会吸收更多Oxonols类染料。 用流式细胞仪测量的方法优点是,灵敏,荧光强度精确定量,快速高通量的检测逐个细胞,可同时检测细胞的多个活性指标,提高可信度,结果具有统计意义。缺点是,实验较MTT等复杂,费用较高。 常用的细胞活性测定方法有台盼蓝染色法、克隆(集落)形成法、3H放射性同位素掺入法、MTT法等。其中MTT法以其快速简便,不需要特殊检测仪器、无放射性同位素、适合大批量检测的特点而得到广泛的应用。但MTT法形成的Formazan为水不溶性的,需要加有机溶剂溶解,由于在去上清操作时会有可能带走小部分的Formazan,故有时重复性略差。为了解决这个问题,研究人员又开发了很多种水溶性的四氮唑盐类:如XTT、CCK-8(WST-8)等。 1.MTT法 MTT:化学名:3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐,商品名:噻唑蓝。检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性MTT还原为水不溶性的蓝紫色结晶甲臜(Formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(DMSO)能溶解细胞中的甲臜,用酶联免疫检测仪在490nm波长处测定其光吸收值,可间接反映活细胞数量。在一定细胞数范围内,MTT结晶形成的量与细胞数成正比。该方法已广泛用于一些生物活性因子的活性检测、大规模的抗肿瘤药物筛选、细胞毒性试验以及肿瘤放射敏感性测定等。它的特点是灵敏度高、经济。 缺点:由于MTT经还原所产生的甲臜产物不溶于水,需被溶解后才能检测。这不仅使工作量增加,也会对实验结果的准确性产生影响,而且溶解甲臜的有机溶剂对实验者也有损害。
激酶检测:PKC蛋白激酶活性检测实验服务案例 实验试剂:琼脂糖(Promega公司,V3125);其他常备试剂购自sigma公司;PKC活性检测试剂盒(Promega公司, V5330) 实验仪器:电泳仪(北京六一厂,112-0640);水平电泳槽(北京六一厂,122-3146);电热恒温培养箱(碧云天生物,EBI025);电热恒温水浴锅(江苏省金坛市环宇科学仪器厂,HH-8) 基本原理:分离PKC蛋白(磷酸化蛋白及非磷酸化蛋白),利用试剂盒组分将2种蛋白分离并加以区分,利用电泳琼脂糖凝胶技术显示2种不同的蛋白,利用分析软件将显示的不同蛋白定量并加以比较,以磷酸化PKC非磷酸化PKC灰度值IOD的比值显示PKC活性(PKC 活化度)。 操作步骤: 1.用预冷的匀浆器把细胞匀浆。 2.在4℃,用14000 × g 离心裂解液5 分钟,保存上清。 3.用PKC 抽提液预平衡1ml 的DEAE 纤维素柱,再把第2 步中得到的上清过柱。用5ml的PKC 抽提液洗柱子,然后用5ml 含有200mM NaCl 的PKC 抽提液洗脱含有PKC 的组分。4.用PKC 稀释液(PKC dilution buffer)将少量蛋白激酶C(Promtein Kinase C)稀释到 2.5ug/ml。随试剂盒所附的产品信息中标有这个酶的初始浓度。 5.用探头超声波破碎仪超声处理PKC Activator Solution 20-30 秒,或直到溶液变温暖。6.对每一个样品,按照下面所列顺序在0.5ml 小离心管中混合PepTag? PKC 5×BufferPepTag? C1 Peptide,超声过的PKC Activator 5X Solution,和水。在样品加入之前,小离心管一直要放在冰上。阴性对照将用于对反应进行定量时确定其摩尔吸收值(说明书第IV.部分)。 标准PKC检测 PepTag? PKC Reaction 5X Buffer,5ul PepTag? C1 Peptide (0.4ug/ul),5ul PKC Activator 5X Solution 超声处理的,5ul 短肽保护液(非必须),1ul 样品, 9ul 去离子水使终体积为,25ul PKC阳性对照检测 PepTag? PKC Reaction 5X Buffer,5ul PepTag? C1 Peptide (0.4ug/ul) ,5ul PKC Activator 5XSolution 超声处理的,5ul 短肽保护液(非必须),1ul
细胞活性测定方法介绍和使用探讨 细胞活性测定方法有台盼蓝染色法、克隆(集落)形成法、3H放射性同位素掺入法、MTT法等。其中MTT法以其快速简便,不需要特殊检测仪器、无放射性同位素、适合大批量检测的特点而得到广泛的应用。但MTT法形成的Formazan为水不溶性的,需要加有机溶剂溶解,由于在去上清操作时会有可能带走小部分的Formazan,故有时重复性略差。为了解决这个问题,研究人员又开发了很多种水溶性的四氮唑盐类:如XTT、CCK-8(WST-8)等。 现就这三种四氮唑盐类方法作一个简单介绍: 1、 MTT 法 MTT:化学名:3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐,商品名:噻唑蓝。检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性MTT还原为水不溶性的蓝紫色结晶甲瓒(Formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(DMSO)能溶解细胞中的甲瓒,用酶联免疫检测仪在490nm波长处测定其光吸收值,可间接反映活细胞数量。在一定细胞数范围内,MTT结晶形成的量与细胞数成正比。该方法已广泛用于一些生物活性因子的活性检测、大规模的抗肿瘤药物筛选、细胞毒性试验以及肿瘤放射敏感性测定等。它的特点是灵敏度高、经济。 缺点:由于MTT经还原所产生的甲瓒产物不溶于水,需被溶解后才能检测。这不仅使工作量增加,也会对实验结果的准确性产生影响,而且溶解甲瓒的有机溶剂对实验者也有损害。 2、 XTT 法 XTT,化学名: 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide,作为线粒体脱氢酶的作用底物,被活细胞还原成水溶性的橙黄色甲瓒产物。当XTT与电子偶合剂(例如PMS)联合应用时,其所产生的水溶性的甲瓒产物的吸光度与活细胞的数量成正比。 优点:1、使用方便,省去了洗涤细胞;2、检测快速;3、灵敏度高,甚至可以测定较低细胞密度;4、重复性优于MTT。 缺点:XTT水溶液不稳定,需要低温保存或现配现用。
抗氧化酶(SOD、POD、CAT)活性测定方法 一、超氧化物歧化酶(SOD)活性测定(氮蓝四唑光化还原法) 1、试剂的配制 (1)0.05mol/L磷酸缓冲液(PBS,pH7.8): A母液:0.2mol/L磷酸氢二钠溶液: 取Na2HPO4·12H2O(分子量358.14)71.7g; B母液:0.2mol/L磷酸二氢钠溶液:取NaH2PO4·2H2O(分子量156.01)31.2g。 分别用蒸馏水定容到1000ml。 0.05mol/L PBS(pH7.8)的配制:分别取A母液(Na2HPO4) 228.75ml,B母液(NaH2PO4) 21.25ml,用蒸馏水定容至1000ml。 参考文献:李合生主编:植物生理生化实验原理和技术.高等教育出版社,2000:267~268。 (2)14.5mM甲硫氨酸溶液:取 2.1637g Met用磷酸缓冲液(pH7.8)定容至1000ml。 (3)30μM EDTA-Na2溶液:取0.001gEDTA-Na2用磷酸缓冲液定容至100ml。 (4)60μM核黄素溶液:取0.0023g核黄素用磷酸缓冲液定容至100ml,避光保存。 (5)2.25mM 氮蓝四唑(NBT)溶液:取0.1840g NBT用PBS定容至100ml,避光保存。 酶液制备:取0.2g(可视情况调整)样品(新鲜叶片或根系)洗净后置于预冷的研钵中, 加入1.6ml 50mmol/L预冷的磷酸缓冲液(pH7.8)在冰浴上研磨成匀浆,转入离心管中在4℃、12000g下离心20min,上清液即为酶液。 2、酶活性测定 (1)反应混合液配制(以60个样为准):分别取Met溶液162ml,EDTA-Na2溶液0.6ml,磷酸缓冲液 5.4ml,NBT溶液6ml,核黄素溶液6ml,混合后摇匀; (2)分别取3ml反应混合液和30μl酶液于试管中 (3)将试管置于光照培养箱中在4000 lux光照下反应20min; 同时做两支对照管,其中1支试管取3ml反应混合液加入30μl PBS(不加酶液)照光 后测定作为最大光还原管,另1支只加缓冲液置于暗中测定时用于调零。 (4)以不照光的对照管(只有缓冲液并置于暗处)调零后,避光测OD560(出现颜色即可测定)。
蛋白激酶 1、按底物蛋白被磷酸化的氨基酸残基种类(5类) ①.丝氨酸/苏氨酸(Ser/Thr)蛋白激酶 ②.酪氨酸(Tyr)蛋白激酶: 1.EGFR(EGFR、HER2/ErbB2、ErbB3、ErbB4):表皮生长因子受体 2.PDGFR(PDGFRα、PDGFRβ):血小板衍生生长因子受体 CSF1R: 集落刺激因子1受体 c-Kit:干细胞生长因子受体 Flk2:胎肝激酶2 3.InsR:胰岛素受体 IGF-1R:类胰岛素生长因子受体 IRR:胰岛素相关受体 4.NGFR:神经生长因子受体 5.FGFR(FGFR1、FGFR2、FGFR3、FGFR4):成纤维细胞生长因子受体 6.VEGFR(VEGFR1、VEGFR2/FLK-1、VEGFR3/FLT4):血管内皮生长因子受体 7.HGFR:肝细胞生长因子受体 8.c-Met Ron sea 9.Ltk Alk 10.c-RET 11.Ros 12.Eph、Eck、Eek、Erk、Elk 13.Tie、Tie-2 Src家族 Tec家族(Btk、Itk/Tsk/Emt、Tec、Txk和Bmx等) ZAP70家族 、TYK1)等 Abl Wee等 ③.组氨酸蛋白激酶(组氨酸、精氨酸或赖氨酸的碱性残基被磷酸化,见于双组分信号系统) ④.色氨酸蛋白激酶 ⑤.天冬氨酰基/谷氨酰基蛋白激酶 2、按序列相似性及功能(7类) ①.AGC组:核苷酸依赖家族(PKA、PKG、PKC家族) ②.CaMK组:Ca2+/钙调素调节的蛋白激酶家族、snfl/AMPK家族 ③.CMGC组:CDK、MAPK、GSK3、CLK家族
④.CKI:酪氨酸激酶家族I ⑤.TK:酪氨酸蛋白激酶 ⑥.TKL:类似酪氨酸激酶 ⑦.STE 癌症 1、癌症主要有四种: 1、癌瘤:影响皮肤、粘膜、腺体及其他器官; 2、血癌:即血液方面的癌; 3、肉瘤:影响肌肉、结缔组织及骨头; 4、淋巴瘤:影响淋巴系统。 常见的癌症有血癌(白血病)、骨癌、淋巴癌(包括淋巴细胞瘤)、肠癌、肝癌、胃癌、盆腔癌(包括子宫癌,宫颈癌)、肺癌(包括纵隔癌)、脑癌、神经癌、乳腺癌、食道癌、肾癌等。
植物组织中SOD活性的测定超氧物歧化酶(SOD)是需氧生物中普遍存在的一种含金属酶。它与过氧化物酶、过氧化氢酶等酶协同作用防御活性氧或其他过氧化物自由基对细胞系统的伤害;SOD可以催化氧自由基的歧化反应,生成过氧化氢、过氧化氢又可以被过氧化氢酶转化成无害的分子氧和水。 【原理】 依据SOD抑制NBT在光下的还原作用来确定酶活性大小。在有氧化物存在下,核黄素可被光还原,被还原的核黄素在有氧的条件下极易再氧化而产生02-,02- 可将NBT还原为蓝色的甲,后者在560 nm 处有最大吸收。而SOD作为氧自由基的清除剂可抑制此反应。于是光还原反应后,反应液蓝色愈深,说明酶活性越低,反正酶活性越高。一个酶活单位定义为将NBT的还原抑制到对照一半(50%)时所需的酶量。 【仪器与用具】 离心机;分光光度计;进样器;紫光灯(4000 Lx);15 mm×150 mm试管;黑色硬质套。 【试剂】 ①0.05mol/L 磷酸缓冲液PBS(pH=7.8) 提取介质50mmol/L PBS(pH=7.8)内含1% 聚乙烯吡咯烷酮 ② 130mmol/L 甲硫氨酸溶液:称1.9397g Met,用PBS 7.8 进行溶解 并定容至100ml;
③ 750umol/L NBT:称0.06132g NBT,用PBS 7.8进行溶解并定容至 100ml,避光保存; ④20umol/核黄素溶液:称0.0075g核黄素,用蒸馏水溶解定容至 1000ml,避光保存,现用现配; ⑤ 100umol/L的EDTA-Na2溶液:称0.0372g EDTA-Na2 ,用PBS 7.8 定容至1000ml。 【方法】 1.酶液提取 称取植物叶片0.2g(去叶脉)于预冷的研钵中,加1ml预冷的提取介质在冰浴下研磨成匀浆,加入提取介质冲洗研钵,并使最终体积为2ml,于4℃、10000r/min离心15min,上清液即为SOD粗提液。 2.显示反应 取透明度好、质地相同的15mm×150mm试管4支,2支为测定,2支为对照,按下表加入试剂。混匀后,给1支对照管罩上比试管稍长的双层黑色硬质套遮光,与其他各管同时置于4000lx日光灯下反应20-30min,反应温度控制在25-35℃ 【计算】
实验试剂:琼脂糖(Promega公司,V3125);其他常备试剂购自sigma公司;PKC活性检测试剂盒(Promega公司, V5330) 实验仪器:电泳仪(北京六一厂,112-0640);水平电泳槽(北京六一厂,122-3146);电热恒温培养箱(碧云天生物,EBI025);电热恒温水浴锅(江苏省金坛市环宇科学仪器厂,HH-8)基本原理:分离PKC蛋白(磷酸化蛋白及非磷酸化蛋白),利用试剂盒组分将2种蛋白分离并加以区分,利用电泳琼脂糖凝胶技术显示2种不同的蛋白,利用分析软件将显示的不同蛋白定量并加以比较,以磷酸化PKC非磷酸化PKC灰度值IOD的比值显示PKC活性(PKC活化度)。 操作步骤: 1.用预冷的匀浆器把细胞匀浆。 2.在4℃,用14,000 × g 离心裂解液5 分钟,保存上清。 3.用PKC 抽提液预平衡1ml 的DEAE 纤维素柱,再把第2 步中得到的上清过柱。用5ml的PKC 抽提液洗柱子,然后用5ml 含有200mM NaCl 的PKC 抽提液洗脱含有PKC 的组分。 4.用PKC 稀释液(PKC dilution buffer)将少量蛋白激酶C(Promtein Kinase C)稀释到2.5ug/ml。随试剂盒所附的产品信息中标有这个酶的初始浓度。 5.用探头超声波破碎仪超声处理PKC Activator Solution 20-30 秒,或直到溶液变温暖。6.对每一个样品,按照下面所列顺序在0.5ml 小离心管中混合PepTag? PKC 5×Buffer,PepTag? C1 Peptide,超声过的PKC Activator 5X Solution,和水。在样品加入之前,小离心管一直要放在冰上。阴性对照将用于对反应进行定量时确定其摩尔吸收值(说明书第IV.部分)。 标准PKC检测 PepTag? PKC Reaction 5X Buffer 5ul PepTag? C1 Peptide (0.4ug/ul) 5ul PKC Activator 5X Solution, 超声处理的 5ul 短肽保护液(非必须) 1ul 样品 9ul 去离子水使终体积为 25ul PKC阳性对照检测 PepTag? PKC Reaction 5X Buffer 5ul PepTag? C1 Peptide (0.4ug/ul) 5ul PKC Activator 5XSolution, 超声处理的 5ul 短肽保护液(非必须) 1ul Protein Kinase C (2.5ug/ml 溶于 PKC dilution buffer) 4ul 去离子水使终体积为 25ul PKC阴性对照检测(不加PKC) PepTag? PKC Reaction 5X Buffer 5ul PepTag? C1 Peptide (0.4ug/ul) 5ul PKC Activator 5XSolution, 超声处理的 5ul 短肽保护液(非必须) 1ul 去离子水使终体积为 25ul
SOD酶活测定方法 (一)氮蓝四唑法 1. 方法提要 在电子供体如甲硫氨酸存在下,核黄素受光激发,与电子供体反应被还原。在氧气中,还原的核黄素与氧化反应产生,将无色(或微黄)的氮蓝四唑还原为蓝色的不溶性僭,SOD 通过催化歧化反应,生成O2与H2O2,从而抑制蓝色形成。按抑制蓝色特形成的50%为一酶活单位。酶活力越高,抑制50%蓝色形成所需酶量越少。 2. 仪器 荧光灯管。 离心机。 分光光度计。 pH计。 3. 试剂 (1)磷酸氢二钾(K2HPO4·3H2O),磷酸二氢钾(KH2PO4),甲硫氨酸(Met),氮蓝四唑(NBT),核黄素,乙二胺四乙酸(EDTA),以上试剂均为分析纯级;所用水为离子水或同等纯度蒸馏水。 (2)pH7.8, 5.0×10-2mol/L的K2HPO4- KH2PO4缓冲液(于冰箱中保存)。 4. 测定步骤 (1)酶液的制备:称取5~10g样品,加预先在冰箱中放置的上述K2HPO4- KH2PO4缓冲液,缓冲液的量为所用样品的10倍以上,在4℃条件下或冰浴中研磨成匀浆,四层纱布过滤,滤液经4000r/min离心20min,取上清液用于酶活测定。 (2)酶反应酬体系液的制备:取上述K2HPO4- KH2PO4缓冲液30ml,依次溶入Met,NBT,核黄素与EDTA,使它们的浓度分别为 1.3×10-2mol/L, 6.3×10-5mol/L, 1.3×10-6mol/L与1×10-4mol/L,放冰箱中避光保存。 (3)测酶活在暗光下,取上述酶反应体系液3mL,移入试管中,试管放在一反应小室中,反应小室壁上贴锡箔纸,应将每个试管摆放在照光后所接受光强一致的位置。向每支试管加入25~30μL酶液。在25~30℃下用光强4000lx的荧光灯管(可用15W荧光灯)进行光照,15~20min后,出现颜色变化。停止光照。在560nm波长下比色测量透光度。用未加酶液的反应体系做对照。 5. 结果计算 以抑制蓝色形成的50%为一个酶活单位。按下式计算: 式中s——样品照光后的透光度; a——未加酶之反应液照光后透光度; b——未加酶之反应液照光前透光度; n——酶液稀释倍数。 样品酶活单位表示:SOD酶活单位(U)/g干重(g鲜重或g蛋白)。 6. 注释 (1)进行照光操作时,应注意所用试管的直径与管壁厚度基本一致。 (2)进行比色测定时,应用未加核黄素的酶反应体系液作空白。 (二)连苯三酚自氧化法 1. 方法提要 连苯三酚在碱性条件下,能迅速自氧化,释放出,生成带色的中间产物。在自氧化过程的初始阶段,黄色中间物的积累在滞后30~45s后就与时间成线性关系。中间产物在420nm处
1、细胞活性的鉴定:死细胞对许多抗体均有很强的非特异性染色,这就使样本细胞活性检测变得非常重要,尤其是经过长时间运输和储存的样本。检测的方法通常有两种:(1)实时的流式检测:利用荧光染料PI、7-AAD或EMA 进行死细胞染色,而活细胞拒染这些染料。此方法的优势是细胞表面标志和活性分析可同时进行。尤其适用于高度坏死的样本。7-AAD最常用,因为在488nm激发下,其最大发射光在670nm左右,适合与FITC或PE进行多色标记。但随着时间延长,7-AAD会在固定的细胞群体重新分配,死活细胞的区分变得困难。因此,对于染色并在固定后12小时以上分析的标本,最好用EMA。EMA与死细胞DNA稳定的共价结合保证了长时间固定后仍能很好的区分固定前的死活状态。(2)手工检测:使用Trypanblue或其他细胞活性染料。(3)使用专门的仪器进行检测。如Vi-cell。 2、7-AAD (7-amino-actinomycin D)是一种核酸染料,在流式细胞术中用于鉴定死细胞,这种作用与PI相似。它不能通过正常质膜,随着细胞凋亡、细胞死亡过程,质膜对7-AAD的通透性逐渐增加,结合细胞凋亡中DNA的有控降解,在合适波长激发光的激发下可发出明亮的红色荧光,通过7-AAD标记DNA的强弱,将细胞分为三群:7-AAD 强为死亡细胞,7-AAD弱是凋亡细胞,7-AAD-为正常活力细胞。7-AAD同PI 有着相似的荧光特性,但其发射波谱较PI窄,PI检测时同时占据FL-2和FL-3通道,而7-AAD只占FL-3通道,对其他检测通道的干扰更小,在多色荧光分析中是PI 的最佳替代品,可与Annexin V-EGFP/FITC/PE联合使用。 3、Annexin V-PE/7-AAD细胞凋亡检测试剂盒是用红色荧光染料PE(Phycoerythrin)标记的AnnexinV作为探针,来检测细胞早期凋亡的发生,可用荧光显微镜、流式细胞仪或其他荧光检测设备进行检测。 其检测原理为:在正常的活细胞中,磷脂酰丝氨酸(phosphotidylserine,PS)位于细胞膜的内侧,但在早期凋亡的细胞中,PS 从细胞膜的内侧翻转到细胞膜的表面,暴露在细胞外环境中。Annexin-Ⅴ(膜联蛋白-V)是一种分子量为35-36KD的Ca2+依赖性磷脂结合蛋白,能与PS高亲和力结合。可通过细胞外侧暴露的磷脂酰丝氨酸与凋亡早期细胞的胞膜结合。因此Annexin V是检测细胞早期凋亡的灵敏指标。 另外,本试剂盒中提供的7-AAD可用来区分存活的早期细胞和坏死或晚期凋亡细胞。7-AAD是一种核酸染料,同PI 有着相似的荧光特性,但其发射光谱较PI窄,对其他检测通道的干扰更小,在多色荧光分析中是PI的最佳替代品,可与Annexin V联合使用。该染料不能透过正常细胞或早期凋亡细胞的完整的细胞膜,但可穿透晚期凋亡细胞或者坏死细胞并与其内的DNA结合。因此将AnnexinV-PE与7-AAD联合使用时,7-AAD则被排除在活细胞(AnnexinV-/7-AAD-)和早期凋亡细胞(AnnexinV+/7-AAD-)之外,而晚期凋亡细胞和坏死细胞同时被AnnexinV-PE 和7-AAD结合染色呈现双阳性(AnnexinV+/7-AAD+),可以将凋亡早期的细胞和晚期的细胞以及死细胞区分开来。。 操作方法 样品染色 1)悬浮细胞:300g,4℃离心5 min收集细胞。 贴壁细胞:用不含EDTA的胰酶消化后,300g,4℃离心5 min收集细胞。胰酶消化时间不宜过长,以防引起假阳性。2)用4℃预冷的PBS洗涤细胞2次,每次均需300g,4℃离心5 min。 3)加入250μl 1×Binding Buffer重悬细胞,调节其浓度为1×106细胞/ml。 4)取100 μl细胞悬液于5ml流式管中,加入5μl Annexin V/PE和10 μl7-AAD,轻轻混匀。 5)避光、室温反应15 min。 6)加入400μl1×Binding Buffer,混匀,样品在1小时内检测。 4、CFSE (carboxyfluoresceindiacetate, succinimidyl ester 羧基荧光素双乙酸盐,琥珀酰亚胺酯) 被动扩散进入细胞,其本身是无色无荧光的,被细胞内的酯酶分解后产生高强度的绿色荧光,定位于细胞膜、细胞质和细胞核,在细胞核的荧光染色最强,该荧光产物与胞内的氨基结合长时间留存于细胞内并可被乙醛固定。未结合的试剂与副产物扩散至胞外基质中,被洗去。 传代或胚胎分裂分析,CFSE与赖氨酸侧链或其他氨基基团反应,共价偶联至细胞内和细胞表面的蛋白,细胞分裂时被平均分配至子代细胞,荧光强度降到母代细胞的一半。适合用于胞内示踪,在细胞分裂或融合后分配至子代细胞中,并不会被转移至邻近的细胞。在小鼠淋巴细胞迁移实验中,CFSE注射进入小鼠体内后,标记的淋巴细胞的检测可长达8周。肝内注射荧光显微镜定位检测可长达20天。
MTT检测细胞活性的操作方法 一、MTT是什么 MTT是一种粉末状化学试剂,全称为3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di-phenytetrazoliumromide,汉语化学名为3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐,商品名:噻唑蓝。是一种黄颜色的染料。 二、MTT法用来做什么 简单地说:是一种检测细胞存活和生长的方法。 MTT主要有两个用途 1.药物(也包括其他处理方式如放射线照射)对体外培养的细胞毒性的测定; 2.细胞增殖及细胞活性测定。 三、为何MTT可以用来做上述工作 检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性MTT还原为水不溶性的蓝紫色结晶甲瓒(Formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(DMSO)能溶解细胞中的甲瓒,用酶标仪在490nm波长处测定其光吸收值,在一定细胞数范围内,MTT结晶形成的量与细胞数成正比。根据测得的吸光度值(OD值),来判断活细胞数量,OD值越大,细胞活性越强(如果是测药物毒性,则表示药物毒性越小)。 四、实验所需材料 1.MTT 溶液的配制通常MTT 配成的终浓度为5mg/ml,须用PBS或生理盐水做溶剂。 市面上一般MTT的包装为100mg,250mg或1g 1.1对于100mg这样的小包装,厂家都是将MTT放入小管中的,个人建议不要再用天平称量分装,而应该一次性将其全配制成溶液,如100mg用20mlPBS来溶解。具体做法:预先在50ml离心管(没有的话,可用培养瓶替代)加入20ml PBS,从中先吸取500-1000ul PBS装入含MTT的小管中,吹打若干次后将其移入50ml离心管,然后再混匀。可以重复几次,以使小管中的MTT不残留于管内。将MTT完全混匀后,用0.22μm滤膜过滤以除去溶液里的细菌,分装避光(避光袋或是黑纸、锡箔纸包住)可长期保存于-20度。按细胞培养板每孔需加10ul计算,一般每96孔板约需1ml,所以分装时可考虑每管分装1ml。 1.2对于大包装,可按上述方法称取一部分溶解,也有人将粉剂分装在EP管里,用的时候现配,直接往培养板中加。 注意事项: 1. 在配制和保存的过程中,容器最好用铝箔纸包住。 配成的MTT需要无菌,MTT对菌很敏感 MTT一般最好现用现配,过滤后4度避光保存两周内(个人曾做过4度避光保存4周的MTT溶液,效果仍然不错)有效,或配制成5mg/ml保存在-20度长期保存,避免反复冻融,最好小剂量分装,用避光袋或是黑纸、锡箔纸包住避光以免分解。当MTT变为灰绿色时就绝对不能再用。 MTT有致癌性,用的时候小心,有条件最好带那种透明的簿膜手套. 2.MTT甲瓒溶解液 2.1二甲基亚砜DMSO,可以直接溶解,无需配制,使用方便,溶解速度快,但对人体毒性较大,且需去除原培养液,在去除培养液的过程中,可能会把结晶去掉,导致结果不稳定。 2.2三联溶解液:SDS10g,异丁醇5ml,10M HCl 0.1ml 用双蒸水溶解配成100ml溶液,该溶解液不需去除原培养基,但溶解较慢。 该溶解液因含有SDS,在低温保存的时候易产生结晶,因此在用之前必须提前几小时