Essential Requirements Checklist of the MDD(07 47 EC)基本要求检查表(中英文)
- 格式:doc
- 大小:280.00 KB
- 文档页数:23

ISO 13485: 2016 Pla nner and Delta ChecklistInstructions:1.Highlighted areas are to be completed by the Client Organization prior to the off-site review, or on-site Gap Analysis or Upgrade Audit, and submitted to the NSF-ISR Lead Auditor for review.2.The Annex A - ISO 13485: 2016 vs. ISO 13485: 2003 Comparison Table has been provided at the end of this document for information and reference purposes only.pletion by the Client Organization should include the final statement of readiness for Upgrade by the Top Management of the Client Organization.4.The columns for aPlanned Completion Date ” and Responsibility ” may be used by the Client Organization to develop their plan upgrading their QMS to the requirements of ISO 13485: 2016.5.All other areas of the Checklist are required to be completed by the N S F-ISR Lead Auditor to confirm the effective implementation of th e Client Organization ' s ISO 13485: 2016ialityManagement System.6.The Lead Auditor shall sign the appropriate sections at the end of the Checklist to indicate: whether the Client Organization is Ready/Not Ready for Upgrade Audit (Off-site review), AND the finalapproval of the QMS in meeting the requirements of ISO 13485: 2016 (during the on-site Upgrade Audit)7.This checklist shall be submitted by the NSF-ISR Lead Auditor as one of the records of the ISO 13485: 2016 Upgrade for the Client Organization.Page 1 of 10NOTE: Please en sure that your Orga ni zati on ' s registered ISO 13485:2003 QMS rema ins complia nt with that versi on of the Standa rd un til the Tran siti on to ISO 13485: 2016 is complete and verified by the NSF-ISR Lead Auditor.。

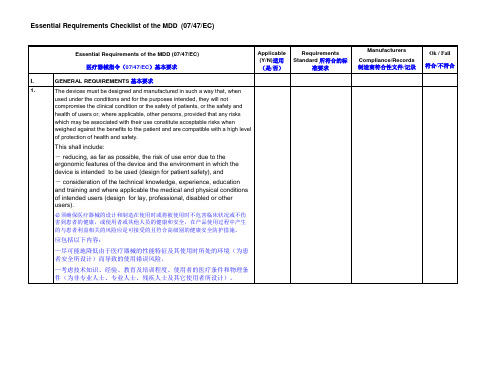
Table of contentsPage 1 Executive Summery 2 1.1 Overview 2 1.2 Commercial Marketing history 2 1.3 Intended uses and indications 2 1.4 List of regulatory approval or marketing clearance obtainedAnd status of any pending request for market clearance 31.5 Important safety/performance related information 32 Relevant Essential Principles and Method Used toDemonstrate Conformity 4 3 Device Description 4 3.1 Device Description and Features 4 3.2 Intended use, Indications, Instructions of Use,Contraindications, Warnings, Precautions, Potential AdverseEffects 4 3.3 Alternative Therapy 4 3.4 Materials 4 3.5 Other Relevant Specifications 53.6 Other Descriptive Information 54 Summary of Design Verification and Validation Documents5 4.1 Sterilization Validation 5 4.2 Stability Studies 5 4.3 Pre-clinical Studies 5 4.3.1 Bio-compatibility Testing 5 4.3.2 Physical Testing6 4.3.3 Animal Studies 6 4.3.4 Software Validation Studies 6 4.3.5 Devices Containing Biological Material 64.4 Clinical Evidence 65 Device Labeling 66 Risk Management 67 Manufacturer Information and Manufacturing Process 6List of AnnexesAnnex:A Regulatory approval certificates and lettersB Declaration of non-FSCAsC Declaration of ConformityD Bio-compatibility ReportsE Essential Requirements checklistF Product Flow ChartG User s’ ManualH Collect of Clinical Data and ReferencesI Sign and labelJ Risk Analysis reportK Design risk assessment reportL Risk Analysis of SoftwareM ISO 13485 certificates1. Executive Summary1.1 OverviewThe ODM-1000 Series Ultrasonic Biometer/Pachymeter is designed to measure the Biological parameters of human eyes. The series consists of three function models: ODM-1000A Ultrasonic Biometer, ODM-1000P Pachymeter and ODM-1000A/P Ultrasonic Biometer. Each model has the following function:ODM-1000A Designed to measure the Depth of Anterior Chamber, Axial Lengthof human eyeball.ODM-1000P Designed to measure the Cornea Thickness.ODM-1000A/P Combination of the two1.2 Commercial Marketing HistoryMEDA Co., Ltd. first began marketing the product in EU countries and other countries. The dates of first introduction and list of countries where the ODM-1000 Series Ultrasonic Biometer is marketed is detailed in Table 1.1.3 Intended uses and indicationsIntended useThe ODM-1000A/P intended used for ophthalmology to accurately measure the axiallength (AL), anterior chamber depth (ACD), lens thickness (LT), and corneal thickness (CT).The ODM-1000A intended used for ophthalmology to accurately measure the axial length (AL), anterior chamber depth (ACD), lens thickness (LT).The ODM-1000P intended used for ophthalmology to accurately measure thecorneal thickness (CT).IndicationsThe ODM-1000 Series Ultrasonic Biometer is indicated for providing basic parameters information of the eyeball before a surgery.1.4 List of Regulatory approval or marketing clearance obtained and status of any pending request for market clearance.Registration StatusThe registration status of the ODM-1000 Series Ultrasonic Biometer in the reference agencies are shown in T able 2. The following regulatory approval certificates and letter are provided in Annex A:♥ US FDA – 510(k) Pre-market notification letter♥TŰV SŰD—Directive 93/42/EEC Annex II, Section 3. CE Certificate♥ Korea—KFDA1.5 Important safety/performance related informationThere is no Adverse Events or Field Safety Corrective Actions (FSCA) for ODM-1000 Series Ultrasonic Biometer. And one declaration on this is attached. See Annex B.2. Essential Principles and Evidence of ConformityThe essential principles conformity checklist is provided in Annex E: Essential Requirements Checklist.3. Device Description3.1 Device Description and FeaturesDescription and principles of operationO DM-1000 A/P Biometer is an ultrasonic measuring instrument of pulse reflection. It contains two independent units: A-Mode Axis Biometric Parameter Measuring Unit and P-Mode Corneal Thickness Measuring Unit. It can be selected by user to configure:ODM-1000A Ultrasonic A-Biometer (only for axial A biometry)ODM-1000P Pachymeter (only for corneal thickness measurement)The A-Biometer consists of a 10MHz A-probe and biometric unit.The axial biometry is the measurement for anterior chamber depth (ACD), lensthickness (LENS), vitreous length (VITR) and axial length (AL).Corneal thickness Pachymeter consists of a 15MHz-pachymetric probe and the measuring unit. It is on the basis of the measurement of time interval between the anterior and post interface reflection wave to get the thickness of the cornea.3.2 Intended use, Indications, Instructions of Use, Contraindications, Warnings, Precautions and Potential Adverse EffectsThe information for these sections is found in Annex G: User’s Manual.3.3 Alternative TherapyThe product is intended for measuring bio-parameters and the same purpose could be achieved by IOL master, which is of laser system.3.4 MaterialsA listing of the components that contacts patients and materials making up theODM-1000 Series Ultrasonic BiometerPlease refer to 4.3.5 of Executive summary (this file).3.5 Other Relevant SpecificationsNot applicable as its specifications already described.3.6 Other Descriptive InformationNot applicable4. Summary of Design Verification and Validation Documents4.1 Sterilization ValidationThis section is not applicable to the ODM-1000 Series Ultrasonic Biometer as it is not provided with any sterilized accessory.4.2 Stability StudiesThis section is not applicable for the ODM-1000 series Ultrasonic Biometer.4.3 Pre-clinical Studies4.3.1 Biocompatibility TestingThe tests as recommended in the ISO 10993-1 (Biological Evaluation of Medical Devices Part 1: Evaluation and Testing) were conducted. All testing was conducted on raw material of the subjected accessories. As summary of biocompatibility test conducted for the ODM-1000 Series Ultrasonic Biometer is found in T able 3.And the detail information could be found in the respective Bio-compatibility Reports that are attached as Annex D: Bio-compatibility Reports.Table 3: Biocompatibility studies conducted4.3.2 Physical TestingThis section is not applicable as the physical characterizations of the housing have no impact on the safety or accuracy.4.3.3 Animal StudiesThis section is not applicable to ODM-1000 Series Ultrasonic Biometer as the clinical trial has been conducted. (See Annex H: Collect of Clinical Data and References)4.3.4 Software Validation StudiesSee Annex L: Risk Analysis of Software.4.3.5 Devices Containing Biological MaterialODM-1000 Series Ultrasonic Biometer contains Biological Material and some accessories provided with the system also contain Biological material. The biological materials could be found in table 4 as below.4.4 Clinical EvidencePlease refer to Annex H: Collect of Clinical Data and References.5. Device LabelingThe device labeling is provided in Annex I: Sign and Label.6. Risk ManagementThe risk management report summarizes the risk management activities completed for the device and is provided in Annex J: Risk Analysis Report. The design and process risk assessments are combined in Annex J: Risk Analysis Report.7. Manufacturer Information and Manufacturing ProcessThe overall manufacturing process flow diagram for the device is shown in Annex F:Product Flow Chart.The whole system is manufactured by an ISO 13485 certified processes that are carried out at Meda Co., Ltd’s manufacturing facility in Tianjin, China.Manufacturer InformationMeda Co., Ltd.Add: C2-3-D, Xinmao Science Skill Park,Huayuan Ind. Dev. Area,Tianjin 300384 ChinaTel: 86-22-83713808-0Fax: 86-22-83713880ISO 13485:2003 certified (Copy of certificate is provided in Annex M)。

说明:在以下答案中,红色问题为范围之外的问题,各位同学可以不用阅读。
蓝色和黑色字体的题目需要给位阅读并背诵。
1. What is quality?The essential character of something, an inherent or distinguishing character, degree or grade of excellence. Quality means, “Meeting requirements.” Whether or not the product or service does what the customer needs. In another word Quality is, “fit for use.”2. Explain “Prevention”&“Detection”Prevention means to prevent quality defects or deficiencies in the first place, and to make the products and processes assessable by quality management program. Prevention decreases production costs because the sooner a defect is located and corrected, the less costly it will be in the long run.The greatest payback is with prevention.3. Explain “Verification”&“Validation”The overall goal of verification is to ensure that each software product developed throughout the software life cycle meets the customer’s needs and objectives as specified in the software requirements document.Validation checks that the system meets the customer’s requirements at the end of the life cycle.Usually verifications take place at the end of each phase. Validations take place just before the product is delivered.It is good practice to combine verification with validation in the testing process.4. Cost of Quality includes _______The cost of quality includes all costs incurred in the pursuit of quality or in performing quality-related activities.The total cost of quality is the sum of four component costs:Prevention cost, appraisal cost, internal failure cost, external failure cost.5. Explain “Prevention Cost” with examplePrevention costs consist of actions taken to prevent defects from occurring the first place.Quality planning, formal technical reviews, test equipment, training …6. Explain “Appraisal Cost” with exampleAppraisal costs consist of measuring, evaluating, and auditing products or services for conformance to standards and specifications.Inspection and testing of products, Equipment calibration and maintenance, Processing and reporting inspection data…7.What are the components of software quality assurance?Software testing, quality control, software configuration management8. What are the elements of software configuration management?Component identification, version control, configuration building, change control.9. Why change control is the key role in the Software ConfigurationManagement?SCM answers Who, What, When, and Why.Who made the changes?What changes were made to the software?When were the changes made?Why were the changes made?10. What are the main responsibilities of SQA group?The SQA group works with the software project during its early stages to establish plans, standards, and procedures that will add value to the software project and satisfy the project constraints and the organization’s policies.The SQA group reviews project tasks and audits software work products throughout the SDLC lifecycle and provides management with visibility as to whether the software project is adhering to its established objectives and standards.11. Why do Dev people think that SQA group arespies tomanagement?12. What is CMMI?Capability Maturity Model Integration——即软件能力成熟度模型集成其目的是帮助软件企业对软件工程过程进行管理和改进,增强开发与改进能力,从而能按时地、不超预算地开发出高质量的软件。


CE技术文件中至少应包含如下内容(有源产品适用)1.封面(如下例,仅供参考)2.目录(可根据自己的文件安排)3.文件更新记录表(如下例,仅供参考)Revision History Sheet4.制造商地址(如公司地址和制造场地地址不同,要写清楚每个地址,如果是经销商,只写明注册地址即可)5.欧盟授权代表(须附与欧盟授权代表签的协议)6.DoC Declaration of Conformity 符合性声明(例,样本见附录1)7.产品名录,产品图片,宣传单等(如有,并请注意宣传册最好加版本号)8.产品/产品族的常规描述8.1产品简介用描述的方式介绍产品或产品族的主要功能,简要的基本工作原理等。
8.2预期用途使用的目的,人群,地点环境等,可参考其它公司同类产品的预期用途来定义8.3产品基本设计实现描述本产品简要的设计原理,可按硬件功能框图或原理框图来稍加描述。
如果是系列产品要说清楚产品族中每个产品之间的相同和不同点。
可列表比较说明。
8.4性能规格(可参照公司的企业标准的技术指标部分,若是系列产品要在指标中体现清楚差异)8.5附件清单表(例如各种配用的探头附件的型号及数量,按产品实际的标配及选配附件填写)8.6分类规则According to 93/42/EEC Annex IX, Rules XX, (产品名) is a class XX device.如一个产品族中分类等级不同要按不同规则来分类8.7认证途径选择Directive MDD93/42/EEC Annex XX,要根据分类的等级来选择合适的认证途径9.适用的产品标准清单列表(例,如下,适用标准应不仅限于上述这些,还应根据本公司具体的产品要求增加需符合的其它欧盟协调标准)需要标列出标准的版本,需为最新版本International Standard & Harmonized European Standard List10.BOM清单产品的材料清单列表,提供可链接到的文件名和文件编号即可。
NBOG’s Best Practice Guideapplicable for AIMDD, ⌧ MDD, and IVDD 2009-4 Guidance on Notified Body‘s Tasks of Technical Documentation Assessment on a Representative Basis1 IntroductionThe Directive 93/42/EEC concerning medical devices (MDD) contains possible conformity assessment procedures in order to CE mark devices. Up to now, there has been inconsistency in the way Notified Bodies (NB) have performed the assessment of technical documentations for Class lla and Class llb products following quality system conformity assessment routes.The Directive 2007/47/EC, covering the revision of the MDD, includes additional requirements for review of technical documentation for all Class IIa and Class IIb medical devices covered by quality system assessment routes.2 ScopeThis guideline has been prepared for NBs on how to assess the technical documentation on a representative basis according to the Directive 2007/47/EC. Attention was given to the sampling size and depth of the assessment. It will also be useful for Designating Authorities in monitoring the performance of their NBs in this area.3 Definitions3.1 Device subcategory (Directive 2007/47/EC, Art. 2, Paragraph 1 (iii), point (l) and Directive93/42 EEC Art 1 Paragraph 2 (l))A set of devices having common areas of intended use or common technology.3.2 Generic device group (Directive 2007/47/EC, Art. 2, Paragraph 1 (iii), point (m) andDirective 93/42 Art. 1 Paragraph 2 (m))A set of devices having the same or similar intended uses or commonality of technology allow-ing them to be classified in a generic manner not reflecting specific characteristics.4 Directive Requirements4.1 Class IIa devicesFor Class IIa devices the requirements are covered in Directive 2007/47/EC at Annex ll, Para-graph 2 (i), Paragraph 5 (f), and Paragraph 6 (e). The Directive 93/42/EEC is amended and An-nex II 7.2, Annex V 6.2 and Annex VI 6.2, states that “For devices in Class IIa the notified body shall assess ... the technical documentation … for at least one representative sample for each device subcategory for compliance with the provisions of this Directive.”4.2 Class IIb devicesFor Class IIb devices the requirements are covered in Directive 2007/47/EC at Annex ll, Para-graph 2 (i). The Directive 93/42/EEC is amended and Annex II 7.3 states that “For devices in Class IIb the notified body shall assess, as part of the assessment in Section 3.3, the technical documentation as described in Section 3.2 (c) for at least one representative sample for each generic device group for compliance with the provisions of this Directive.”4.3 Concept of the reviewsDirective 2007/47/EC, Recital (22) introduces the concept of the reviews of Class lla and llb de-vices: “… The depth and extent of this review should be commensurate with the classification of the device, the novelty of the intended treatment, the degree of intervention, the novelty of the technology or construction materials, and the complexity of the design and/or technology. This review can be achieved by taking a representative example of design documentation of one or more type(s) of devices from those being manufactured. Further review(s), and in particular the assessment of changes to the design that could affect conformity with the essential require-ments, should be part of the surveillance activities of the notified body.”Directive 93/42/EEC is amended and in Annex II 7.4, Annex V 6.3 and Annex VI 6.3, there are rationales provided for the sampling: “In choosing representative sample(s) the notified body shall take into account the novelty of the technology, similarities in design, technology, manu-facturing and sterilisation methods, the intended use and the results of any previous relevant assessments (e.g. with regard to physical, chemical or biological properties) that have been carried out in accordance with this Directive. The notified body shall document and keep avail-able to the competent authority its rationale for the sample(s) taken.”4.4 Requirements for surveillance assessmentsDirective 2007/47/EC Annex ll, Paragraph 2 (i), Paragraph 5 (f), Paragraph 6 (e) and Para-graph 6.4 introduces requirements for surveillance assessments. The Directive 93/42/EEC is amended and in Annex II 7.5, Annex V 6.4 and Annex VI 6.4 it is stated: “Further samples shall be assessed by the notified body as part of the surveillance assessment...”.5 TimingDirective 2007/47/EC Article 4 states Member States “… shall apply those measures from 21 March 2010”.Following the adoption of Directive 2007/47/EC, from 21 March 2010 at the latest, for Class IIb devices following the Annex II assessment route and Class IIa devices following the Annex II, V or VI assessment route there is a specific requirement for technical documentation to be reviewed by the NB on a sample basis. At any rate and at the latest, the first surveillance fol-lowing 21 March 2010 will need to address this element.After the 21 March 2010 new certificates can be issued only according to the new requirements. For existing certificates issued prior to this date please see “Interpretative document of the Commission's services Implementation of Directive 2007/47/EC amending Directives 90/385/EEC, 93/42/EEC and 98/8/EC” [1] for details. In this document the Commission’s ser-vices call upon the member states to require Notified Bodies to ensure that surveillance audits are performed at least once a year as specified in the standard EN ISO /IEC 17021 [2].6 Depth of AssessmentThe assessment of each set of technical documentation selected, as a minimum, should con-sist of a review of– the intended use including confirmation that the product is a medical device and it’s correct classification,– the validity of the essential requirements checklist, especially when harmonized standards have not been applied in full,– risk management file,– pre-clinical data (studies in animal models, biocompatibility, technical performance tests etc.),– clinical evaluation in accordance with 93/42/EEC Annex X (Note the NB review should be as per MEDDEV 2.7.1 [3]),– information supplied by the manufacturer (label, instructions for use),– declaration of conformity or the draft thereof,– other technical documentation based on risk,– and should be done according to the respective parts of NBOG BPG 2009-1 [4]. Consequently, there will be one part of the assessment performed within the normal course of the audit but in addition, file reviews will be necessary.The time spent on the review and the level of expertise used should be proportionate to the risk and complexity of the devices in question. For the more complex devices this may require a detailed technical review. If such an in depth assessment is not considered necessary then a justification should be available.Depending on the risk and complexity of the device(s), different experts may be involved. Therefore, NBs may not be able to perform the complete assessment on site.7 ReportingFor those elements covered the report produced for each technical documentation reviewed should be in line with the NBOG guidance on Design Dossier Report Content [4]. This could be either as part of the audit report or separately as a supplement.Nonconformities/Corrective Action Requests should be raised in the normal manner, including those raised against technical documentation under conformity assessment procedures according to Annex V or Annex VI.8 SamplingFor initial audits of devices in Class IIb the Directive states that the NB shall assess the tech-nical documentation for at least one representative sample for each generic device group (i.e. a set of devices having the same or similar intended uses or commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics). This is the same description as for “generic device groups” in EN ISO 15225 [5], which specifies the GMDN system.For initial audits of devices in Class IIa the Directive states that the NB shall assess the tech-nical documentation for at least one representative sample for each device subcategory (i.e. a set of devices having common areas of intended use or common technology). Since no similar description exists for subcategory/ies like for “generic device groups”, the grouped collectiveterms described in NBOG BPG 2009-3 [6], which define scope expressions for NBs, could be used for this.However, in practice it is expected that the NB identifies the complete range of Class IIa and IIb products manufactured and define a suitable sampling plan for initial audits. This should be done based on the numbers of generic device groups and grouped collective terms.It is considered that if large numbers of samples are required to be taken then the following plan should be used:For Class IIa devices: a sample of each group of scope expressions according to NBOG BPG 2009-3 [6].For Class IIb devices: a sample of generic device groups, referenced by different GMDN preferred terms, to the following plan:Up to 2 groups: a sample from each groupUp to 10 groups: a sample from 3 of these groupsUp to 20 groups: a sample from 5 of these groupsUp to 30 groups: from 7 groups a sampleN > 30 groups: from N/10 + 5 groups a sampleFor surveillance audits the NB should identify the complete range of Class IIa and IIb devices and group them according to preferred terms and grouped collective terms (scope expressions). For each certification period, and at least every 3 years, the NB should identify the technical documentations to be reviewed in line with the above sampling plan. As long as there is still a technical documentation not yet reviewed, at least one technical documentation should be sam-pled each year. This activity may or may not be done in conjunction with routine surveillance assessments. During subsequent certification periods appropriate plans should be drawn up to ensure that in the long run the majority, if not all, of the technical documentations will be assessed.9 Changes/New DevicesManufacturers introducing a new subcategory of Class IIa device or a new generic Class IIb device need to inform their NB who need to review relevant technical documentation prior to the products being placed on the market. For changes to existing designs the normal change requirements apply.Reference Directive 93/42/EEC Annex II, V and VIDirective 2007/47/ECSources [1][2] EN ISO/IEC 17021 : 2006 Conformity assessment – Requirementsfor bodies providing audit and certification of management systems[3][4][5] EN ISO 15225 : 2000/A1 : 2004 Nomenclature – Specification for anomenclature system for medical devices for the purpose of regula-tory data exchange[6]Devices Assessmentsdevice subcategory, generic device group, sam-assessment,Keywords conformitypling basis, technical documentationDate of issue July 2009。
Your Paper’s Title Starts Here: Please CenterFULL First Author1, a, FULL Second Author2'6 and Last Author3,0!Full address of first author, including country一Full address of second author,including country3List all distinct addresses in the same waya email,b email,c emailAbstract: This template explains and demonstrates how to prepare your camera-ready paper for Trans Tech Publications. The best is to read these instructions and follow the outline of this text. Please make the page settings of your word processor to A4 format (21 x 29,7 cm or 8 x 11 inches); with the margins: bottom 1.5 cm (0.59 in) and top 2.5 cm (0.98 in),right/left margins must be 2 cm (0.78 in). (We shall be able to publish your paper in electronic form on our web page ,if the paper format and the margins are correct. If not,we will have to scan your paper which, when compared with an electronic version, results in very poor quality)• Your manuscript will be reduced by approximately 20% by the publisher. Please keep this in mind when designing your figures and tables etc.Keywords: List the keywords covered in your paper. These keywords will also be used by the publisher to produce a keyword index.IntroductionAll manuscripts must be in English,also the table and figure texts, otherwise we cannot publish your paper.Please keep a second copy of your manuscript in your office. When receiving the paper,we assume that the corresponding authors grant us the copyright to use the paper for the book or journal in question. Should authors use tables or figures from other Publications, they must ask the corresponding publishers to grant them the right to publish this material in their paper.Use italic for emphasizing a word or phrase. Do not use boldface typing or capital letters except for section headings (cf. remarks on section headings,below).Organization of the TextSection Headings. The section headings are in boldface capital and lowercase letters. Second level headings are typed as part of the succeeding paragraph (like the subsection heading of this paragraph).Page Numbers. Do not number your paper:Tables. Tables (refer with: Table 1,Table 2,…)should be presented as part of thetext,but in such a way as to avoid confusion with the text. A descriptive title should be placed above each table. Units in tables should be given in square brackets [meV]. If square brackets are not available,use curly {meV} or standard brackets (meV).Special Signs, for example , a y |i £1 () > 士搴r {112 0} should always bewritten in with the fonts Times New Roman or Arial,especially also in the figures and tables.Macros. Do not use any macros for the figures and tables. (We will not be able to convert such papers into our system)Language. All text,figures and tables must be in English.Figures. Figures (refer with: Fig. 1, Fig. 2, •") also should be presented as part of the text,leaving enough space so that the caption will not be confused with the text. The caption should be self-contained and placed below or beside the figure. Generally,only original drawings or photographic reproductions are acceptable. Only very good photocopies are acceptable. Utmost care must be taken to insert the figures in correct alignment with the text.Half-tone pictures should be in the form of glossy prints. If possible,please include your figures as graphic images in the electronic version. For best quality the pictures should have a resolution of 300 dpi(dots per inch).Color figures are welcome for the online version of the journal. Generally,these figures will be reduced to black and white for the print version. The author should indicate on the checklist if he wishes to have them printed in full color and make the necessary payments in advance.Equations. Equations (refer with: Eq. 1,Eq. 2, •••)should be in dented 5 mm (0.2’’). There should be one line of space above the equation and one line of space below it before the text continues. The equations have to be numbered sequentially,and the number put in parentheses at the right-hand edge of the text. Equations should be punctuated as if they were an ordinary part of the text. Punctuation appears after the equation but before the equation number,e.g.Literature ReferencesReferences are cited in the text just by square brackets [1]. (If square brackets are not available, slashes may be used instead,e.g. /2/.) Two or more references at a time may be put in one set of brackets [3,4]. The references are to be numbered in the order in which they are cited in the text and are to be listed at the end of the contribution under a heading References, see our example below.ConclusionIf you follow the “checklist” your paper will conform to the requirements of thepublisher and facilitate a problem-free publication process.References[1]Dj.M. Marie, P.F. Meier and S.K. Estreicher: Mater. Sci. Forum VoL 83-87 (1992),p. 119[2]M.A. Green: High Efficiency Silicon Solar Cells(Trans Tech Publications,Switzerland 1987).[3]Y. Mishing,in: Diffusion Processes in Advanced Technological Materials, edtiedby D. Gupta Noyes Publications/William Andrew Publising,Norwich, NY(2004), in press.[4]G. Henkelman,G.Johannesson and H. Jonsson, in: Theoretical Methods inCondencsed Phase Chemistry, edited by S.D. Schwartz,volume 5 of Progress in Theoretical Chemistry and Physics,chapter,10,Kluwer Academic Publishers (2000).[5]R.J. Ong, J.T. Dawley and P.G. Clem: submitted to Journal of Materials Research(2003)[6]P.G. Clem, M. Rodriguez, J.A. Voigt and C.S. Ashley, U.S. Patent 6,231,666.(2001)[7]Information on 。
剑桥雅思听力住宿类话题住宿是雅思听力中常见的话题之一。
以下是一些可能涉及到的问题和答案。
1. What types of accommodation are available for students?- Students have various options when it comes to accommodation, such as student dormitories, apartments, homestays, and shared houses.2. What are the advantages of living in a student dormitory?- Living in a student dormitory allows students to be close to campus and have easy access to facilities such as the library and sports centers. It also provides opportunities to socialize and interact with other students.3. What are the advantages of renting an apartment?- Renting an apartment provides more independence and privacy compared to living in a dormitory. It also allows students to choose their own roommates and have more control over their living environment.4. What is a homestay?- A homestay is when a student lives with a local family in their home. This can provide a cultural immersion experience and an opportunity to practice the language spoken in the country.5. What are some factors to consider when choosing accommodation?- Factors to consider include location, cost, proximity to campus orpublic transportation, amenities, safety, and the type of living environment desired (such as quiet or social).6. How can students find suitable accommodation?- Students can contact their institution for assistance and recommendations. They can also search online rental websites, join social media groups or forums for housing advertisements, or seek the help of real estate agents.7. Are there any special requirements or restrictions for certain types of accommodation?- Some accommodation options, such as student dormitories or homestays, may have age restrictions or specific rules and regulations. It is important for students to inquire about these requirements before making a decision.8. How can students ensure a smooth transition when moving intoa new accommodation?- Students can make a checklist of things they need to bring, communicate with the landlord or host family about move-in dates and procedures, and familiarize themselves with the new surroundings before moving in.。