地中海贫血临床路径
- 格式:doc
- 大小:55.50 KB
- 文档页数:5


你对地中海贫血了解多少地中海贫血,相信很多人对这个病都不陌生,尤其是在我国南方地区。但是关于这个病你又了解多少呢?一、什么是地中海贫血?地中海贫血(简称地贫)最初发现于地中海区域并因此而得名,也叫“海洋性贫血”。在我国,地贫最多见于广东、广西、四川、贵州等地,其次是长江以南各省市,随着社会经济发展,近年来人口流动和南北通婚日益增多,北方地区地贫基因携带者亦逐渐增多,地贫携带者及患者不再只局限于南方地区。
在医学上它还有一个名称叫“珠蛋白生成障碍性贫血”,看着这个名字就知道“地贫”是因为珠蛋白出了问题而引起的。先来简单说说珠蛋白吧,两条α珠蛋白链和两条β珠蛋白链就组成了正常成人的血红蛋白,就像一辆货车有四壁可以装货运输。如果因机体某个或多个珠蛋白基因异常引起珠蛋白链合成障碍,就会使血红蛋白这辆货车出现问题不能发挥正常功能而导致贫血、溶血等发生,引起成不同程度的贫血。根据受影响珠蛋白链的不同,可以分为α-地贫、β-地贫、γ-地贫、δ-地贫等多种类型,以α地中海贫血和β地中海贫血两类最常见。二、怎么就得了地中海贫血?地贫是常染色体单基因隐性遗传病,由携带地贫基因的父母先天遗传而来。正常人自父母双方各继承2个α珠蛋白基因和1个β珠蛋白基因,因此每人共4个α珠蛋白基因和2个β珠蛋白基因。根据前面所提到的,这6个珠蛋白基因中,不论α还是β,只要1个有问题就会导致“地贫”的发生,所以有“地贫”基因的父母生孩子就像买彩票一样,如果父母并没有给孩子异常的基因,那么这个幸运儿就会是一个完全正常的小孩;如果父母给了少量的异常基因,孩子可能只是携带或者轻度地贫;要是不幸给了较多的异常基因,那么就很有可能是个重型的地贫患儿了。倘若父母都没有“地贫”基因,那小孩也自然无从获得异常的“地贫”基因了。举个例子:假如父母中一人为携带者,另一人为正常人,他们的小孩可能为携带者也可能正常,机率各为50%;若父母两人均为同型地贫基因携带者,那就有可能生出重型地贫的小孩了。三、如何知道自己及家人有没有得地中海贫血?既然“地贫”可能完全没有异常表现,而又有遗传的风险,那明确自己和家人有没有“地贫”就非常重要了。不论有没有异常的表现,“地贫”都是可以通过常规检验表现出来的。常用的检查方法主要有以下一些:1、血常规:这是最常规、最经济、也最方便的检查,常规的婚检、孕产检都离不开它,但缺点就是只能作为最粗略的筛查,不能依据血常规来明确“地贫”诊断。拿到血常规要怎么看报告呢?首先看血红蛋白(Hb),“地贫”患者的血红蛋白可以正常也可以减少(贫血),但要注意,血红蛋白降低仅提示贫血,不能表明贫血的原因;其次,要看红细胞体积(MCV)、平均红细胞血红蛋白含量(MCH)及平均红细胞血红蛋白浓度(MCHC),在地贫患者中,无论其血红蛋白正常与否,这三项指标均会显著降低。因此,若看到血常规结果中上述三项明显低于参考值,就要警惕有无“地贫”的可能性了,这时就需要进一步求助于专科医生鉴别诊断。但要特殊强调的是,临床并不能依靠单凭MCV、MCH及MCHC这三个指标降低就确诊“地贫”,因为在其他一些贫血中也会表现为降低(如缺铁性贫血)。2、血红蛋白电泳:普通的血红蛋白电泳有三项指标,HbA、HbF及HbA2。意义如下:静止型基因携带者及α地贫者血红蛋白电泳HbA2可能正常或降低。β地贫HbA2可能增高。3、基因诊断:这是地中海贫血的确诊手段,可以明确“地贫”的基因类型,但缺点是费用较贵。四、为什么要重视地中海贫血?调查数据显示,全世界每年有超过5 万名地贫患儿出生。

地中海贫血科普文地中海贫血是一种常见的遗传性疾病,特别多见于地中海沿岸地区,故得名地中海贫血。
它是由于基因突变导致红细胞内的血红蛋白合成障碍,从而使红细胞寿命缩短造成贫血。
本文将详细介绍地中海贫血的病因、症状、诊断和治疗等方面的内容。
1. 病因地中海贫血是一种由于β-地中海贫血基因突变导致的遗传性疾病。
在正常情况下,β-地中海贫血基因编码的β-地中海贫血基因产物β-地中海贫血链在红细胞内参与血红蛋白合成。
而地中海贫血患者的β-地中海贫血基因发生突变,导致β-地中海贫血链合成障碍,使血红蛋白含量降低,红细胞寿命缩短,进而导致贫血。
2. 症状地中海贫血的症状主要包括贫血、疲劳、头晕、心悸、气促、肝脏和脾脏肿大等。
患者通常会出现周期性的溶血危机和缺铁性贫血。
溶血危机是地中海贫血最突出的临床表现之一,患者在感染、寒冷、氧气不足或剧烈运动等刺激下,红细胞发生大量溶解,导致贫血加重,出现黄疸、脸色苍白、体力下降等症状。
缺铁性贫血是由于长期缺乏血红蛋白合成所需的铁元素而引起的,主要表现为面色苍白、乏力、食欲不振、头晕等。
3. 诊断地中海贫血的诊断主要依靠临床症状、体格检查和实验室检查等手段。
血液学检查可以发现红细胞总数减少,红细胞体积变小,血红蛋白含量降低,红细胞形态异常等。
进一步的实验室检查如血清铁、总铁结合力、骨髓穿刺等可以用于区分地中海贫血和其他类型的贫血。
4. 预防和治疗对于地中海贫血,目前尚无完全治愈的方法,治疗主要是通过缓解症状和预防并发症。
具体治疗方法包括输血治疗、脾切除术、造血干细胞移植等。
输血治疗是目前最常用的治疗方法,通过输注正常的红细胞来补充患者体内缺乏的健康红细胞。
然而,长期输血治疗会导致铁过载,因此需要进行铁螯合剂治疗来清除体内多余的铁元素。
脾切除术是一种常见的手术治疗方法,通过切除脾脏来减少红细胞的破坏和溶解。
脾脏是溶血危机的重要场所,脾功能亢进的患者容易发生溶血危机,因此脾切除术可以有效减少溶血危机的发生。


地中海贫血是怎么引起的地中海贫血的病因珠蛋白链的分子结构及合成是由基因决定的。
γ、δ、ε和β珠蛋白基因组成“β基因族〞,ζ和α珠蛋白组成“α基因族〞。
正常人自父母双方各继承2个α珠蛋白基因(αα/αα)合成足够的α珠蛋白链;自父母双方各继承1个β珠蛋白基因合成足够的β珠蛋白链。
由于珠蛋白基因的缺失或点突变,肽链合成障碍导致发病。
地中海贫血分为α型、β型、δβ型和δ型4种,其中以β和α地中海贫血较为常见。
1.β珠蛋白生成障碍性贫血(β地中海贫血)β珠蛋白生成障碍性贫血(简称β地贫)的发生的分子病理相当复杂,有100种以上的β基因突变,主要是由于基因的点突变,少数为基因缺失。
2.α珠蛋白生成障碍性贫血(α地中海贫血)大多数α珠蛋白生成障碍性贫血(地中海贫血)(简称α地贫)是由于α珠蛋白基因的缺失所致,少数由基因点突变造成。
白基因的缺失所致,少数由基因点突变造成。
地中海贫血的病症根据病情轻重的不同,分为以下3型。
1.重型出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型的表现是臀状头,长骨可骨折。
骨骼改变是骨髓造血功能亢进、骨髓腔变宽、皮质变薄所致。
少数患者在肋骨及脊椎之间发生胸腔肿块,亦可见胆石症、下肢溃疡。
2.中间型轻度至中度贫血,患者大多可存活至成年。
3.轻型轻度贫血或无病症,一般在调查家族史时发现。
地中海贫血的检查1.β珠蛋白生成障碍性贫血(地中海贫血)(1)重型外周血象呈小细胞低色素性贫血,红细胞大小不等,中央浅染区扩大,出现异形、靶形、碎片红细胞和有核红细胞、点彩红细胞、嗜多染性红细胞、豪-周氏小体等;网织红细胞正常或增高。
骨髓象呈红细胞系统增生明显活泼,以中、晚幼红细胞占多数,成熟红细胞改变与外周血相同。
红细胞渗透脆性明显减低。
HbF含量明显增高,大多>0.40,这是诊断重型β地贫的重要依据。
颅骨某线片可见颅骨内外板变薄,板障增宽,在骨皮质间出现垂直短发样骨刺。

地贫诊断标准地贫(Thalassemia)是一种常见的遗传性血液病,主要表现为红细胞数量减少和血红蛋白合成障碍。
地贫患者常常出现贫血、乏力、黄疸等症状,严重的地贫甚至会影响患者的生活质量和寿命。
因此,及早发现和诊断地贫对于患者的治疗和管理至关重要。
本文将介绍地贫的诊断标准,帮助医生和患者更好地了解和诊断地贫症。
一、临床表现。
地贫患者的临床表现主要包括贫血、乏力、黄疸、脾大等症状。
贫血是地贫最常见的表现,患者常常出现面色苍白、乏力、头晕等症状。
黄疸是由于溶血引起的胆红素沉积过多所致,患者的眼睛和皮肤常常呈现黄色。
脾大是由于红细胞破坏过多导致脾脏肿大,可通过体格检查发现。
二、实验室检查。
1. 血常规,地贫患者的血红蛋白水平常常降低,红细胞数量减少,红细胞平均体积(MCV)和平均血红蛋白含量(MCH)降低。
2. 血涂片检查,可见红细胞形态异常,出现不规则形状、变形等。
3. 血红蛋白电泳,通过血红蛋白电泳可以明确地贫的类型,包括α地中海贫血、β地中海贫血等。
三、遗传学检查。
地贫是一种遗传性疾病,因此遗传学检查对于诊断地贫至关重要。
通过进行α、β地中海贫血基因检测,可以明确患者的基因型,帮助医生进行更准确的诊断和治疗。
四、鉴别诊断。
地贫的临床表现和实验室检查与其他贫血症状有时相似,需要与其他贫血症进行鉴别诊断。
例如,与缺铁性贫血、再生障碍性贫血、溶血性贫血等进行鉴别诊断,以明确诊断和制定治疗方案。
五、诊断标准。
根据临床表现、实验室检查和遗传学检查,结合患者的家族史和病史,可以进行地贫的诊断。
一般来说,符合以下条件可以诊断为地贫:1. 临床表现典型,如贫血、乏力、黄疸等;2. 血常规检查显示血红蛋白水平降低,红细胞数量减少,MCV 和MCH降低;3. 血涂片检查显示红细胞形态异常;4. 血红蛋白电泳明确为地贫类型;5. 遗传学检查显示患者携带地贫相关基因。
总之,地贫的诊断需要综合临床表现、实验室检查和遗传学检查,通过明确的诊断标准可以帮助医生更准确地诊断地贫症,为患者制定合理的治疗方案和管理措施提供依据。

卫办医政发〔2011〕76号文件附件骨髓增生异常综合征-难治性贫血伴原始细胞过多(MDS-RAEB)临床路径(2011年版)一、骨髓增生异常综合征-难治性贫血伴原始细胞过多(MDS-RAEB)临床路径标准住院流程(一)适用对象。
第一诊断为MDS-RAEB(ICD:D46.201)。
(二)诊断依据。
根据《血液病诊断和疗效标准》(张之南、沈悌主编,科学出版社,2008年,第三版)、《World Health Organization Classification of Tumors. Pathology and Genetic of Tumors of Haematopoietic and Lymphoid Tissue.》(2008)、NCCN clinical practice guidelines in oncology: myelodysplastic syndromes(V.2.2010)。
诊断标准:1.RAEB-Ⅰ:(1)外周血:①血细胞减少;②原始细胞<5%;③无Auer小体;④单核细胞<l×l09/L。
(2)骨髓:①1系或多系发育异常;②原始细胞5%–9%;③无Auer小体。
2.RAEB-Ⅱ(1)外周血:①血细胞减少;②原始细胞5%–19%;③有或无Auer小体;④单核细胞<l×l09/L。
(2)骨髓:①1系或多系发育异常;②原始细胞10%–19%;③有或无Auer小体。
(三)治疗方案的选择。
根据《邓家栋临床血液学》(邓家栋主编,上海科学技术出版社,2001年,第一版)、《内科学》(叶任高、陆再英主编,人民卫生出版社)、《内科学》(王吉耀主编,人民卫生出版社,2010,第二版)、NCCN clinical practice guidelines in oncology:myelodysplastic syndromes(V.2.2010)。
首先进行诊断分型,然后根据MDS国际预后积分系统(IPSS)(见表1)进行预后分组。


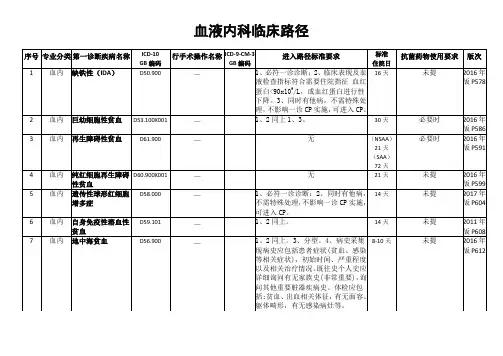
地中海贫血临床路径一、临床路径标准住院流程1、使用对象第一诊断为地中海贫血(ICD: D56. 900)2、诊断依据根据《儿科学》(王卫平主编,人民卫生出版社,全国高等学校教材第8版)、《诸福棠实用儿科学,第七版》(人民卫生出版社)1)病史:面色苍白、乏力、头晕;2)体征:轻度或中度或重度贫血貌;3)实验室检查:血常规:红细胞、血红蛋白、红细胞压积、红细胞平均体积、红细胞平均血红蛋白、红细胞平均血红蛋白浓度下降;4)既往被明确诊断地中海贫血。
3、治疗方案的选择根据《儿科学》(王卫平主编,人民卫生出版社,全国高等学校教材第8版)、《诸福棠实用儿科学,第七版》(人民卫生出版社)1)输血;2)对症支持治疗及根据实验室检查结果对症治疗。
4、标准治疗日为2-15天5、进入路径标准1)第一诊断必须符合地中海贫血(ICD: D56. 900)2)当患者同时具有其他疾病诊断,但在住院期间不需要特殊处理, 也不影响第一诊断的临床路径流程实施6、入院后第一2天1)必须的检查项目:a血常规+血型、尿常规、大便常规;b电解质+肾功能+心肌酶+CRP、肝功能;C输血前四项2)根据患儿病情可选择:血气分析、心电图、腹部B超、影像学检查。
7、药物选择对症治疗:输红细胞8、出院标准1)贫血貌基本纠正;2)复查血常规血红蛋白上升至正常9、变异及原因分析地中海贫血患儿住院经输血治疗但因贫血较重出现心力衰竭等严重危及生命的重症时,应当及时退出地中海贫血临床路径。
临床路径表单使用对象:第一诊断为地中海贫血(ICD: D56.900)页码:1/2 患者姓名:性别:年龄:门诊号:住院号:入径日期:出径日期:入径天数:标准住院日:2-15天使用对象:第一诊断为地中海贫血(ICD: D56.900)页码:2/2患者姓名:性别:年龄:门诊号:住院号:入径日期:出径日期:入径天数:标准住院日:2-15天三、临床路径医嘱明细四、临床路径护理明细。

地中海贫血临床路径一、地中海贫血临床路径标准住院流程(一)适用对象第一诊断为地中海贫血(ICD10:D56.0/D56.100/D56.102/D56.900)(二)诊断依据。
根据《血液病诊断和疗效标准》(张之南、沈悌主编,科学出版社,2008年,第三版)、《诸福棠实用儿科学(第七版)》(人民卫生出版社)、《临床诊疗指南–血液病学分册》(中华医学会编著,人民卫生出版社)。
1.有贫血、黄疸、肝脾肿大、发育不良、“地贫外貌”等临床表现。
2.血象轻型患者Hb常在80g/L以上,重型患者Hb<60g/l,呈小细胞低色素性贫血,红细胞形态不一,大小不均,可见靶形红细胞,网织红细胞增多。
3.骨髓象呈溶血性贫血骨髓象,红细胞系极度增生。
4.血红蛋白分析轻型HbA2 >3.5%,HbF正常或轻度增加(不超过5%),重型HbF增多,多在30%以上,少数HbF为10%~30%,HbA2多正常。
5.家系调查重型患者父母均为β地中海贫血杂合子。
轻型患者父母中一方为β地中海贫血杂合子。
中间型患者父母均为β地中海贫血杂合子;或父母中一方为β地中海贫血杂合子,但其中一方HbF持续存在;或父母中一方为β地中海贫血杂合子,而另一方为aβ地中海贫血。
6.轻型患者应除外缺铁性贫血。
重型及中间型患者应除外HbF增加的其他类型地中海贫血。
(三)治疗方案的选择。
根据《诸福棠实用儿科学(第七版)》(人民卫生出版社)、《临床诊疗指南–血液病学分册》(中华医学会编著,人民卫生出版社)。
(四)标准住院日为14天内。
(五)进入路径标准。
1.第一诊断必须符合ICD10:D56.0/D56.100/D56.102/D56.900地中海贫血疾病编码。
2.血液检查指标符合需要住院指征:血红蛋白<70g/L,或伴有明显缺氧症状,或血红蛋白下降过快。
3.当患者同时具有其他疾病诊断,但在住院期间不需要特殊处理,也不影响第一诊断的临床路径流程实施时,可以进入路径。
各级医疗机构医院地中海贫血分级诊疗流程(2019年版)地中海贫血(地贫)是一种遗传性溶血性贫血病,主要有α和β地贫两种。
珠蛋白链生产不平衡导致α或β链明显过剩,多余的α或β链形成四聚体沉积在红细胞膜上,导致红细胞缺陷,根据缺陷程度的不同出现血管内溶血(α地贫为主)和骨髓内溶血(β地贫为主),即无效造血,这些病理生理最终导致患者出现特殊的贫血,地贫面容(颧骨突出、眼距增宽、鼻梁低平)和肝脾肿大等症状体征。
地贫的治疗主要包括:1.输血与祛铁;2.造血干细胞移植;3.药物治疗;4.基因治疗。
后两者目前正在研究中,有效性和安全性有待证实。
造血干细胞移植(HSCT)治疗地贫已有三十多年的历史,是目前唯一能“根治”地贫和性价比最高的治疗方法。
目前随着移植技术不断进步及移植经验的积累,地贫移植的临床结果变得更好,移植相关并发症及死亡率明显下降。
而且,移植供者也发展到非亲缘供者和HLA不全合亲缘供者;干细胞来源包含骨髓、外周血干细胞,脐血和混合干细胞(如骨髓+外周血干细胞、脐血+骨髓等)移植。
在有经验的中心,同胞全相合移植无地贫生存率(TFS)及总生存率甚至能达到90%左右。
地贫患者符合以下条件者可行HSCT:1.基因诊断确诊为β-地贫纯合子或双重杂合子,临床有中重度贫血并需规律输血才能维持Hb>90g/L者;2. 对依赖输血的其他类型地贫如血红蛋白E(Hb E)病、血红蛋白H(Hb H)病、β地贫复合Hb E等,也可根据实际情况开展。
3.有合适供者;4.知情同意。
以下情况如有合适供者且身体条件符合,在充分知情同意后也可考虑HSCT :依赖长期输血的患者携带罕见基因或其他类型基因变异,无法使用目前基因技术确诊,这类患者在明确了血红蛋白肽链/珠蛋白链异常/非免疫性溶血性贫血诊断后。
一级医院接诊患者时,询问病史,发现Hb及MCV低于正常时;注意缺铁性贫血与地贫鉴别。
难以鉴别时,转诊到上级医院。
二级医院接诊患者时,应评估病情,完善相关实验室检查(血常规、血红蛋白电泳等)。
地中海贫血的临床诊治(上)广东省妇幼保健院产前诊断中心吴箐我们来讨论一下地中海贫血的诊断和治疗的一些相关问题。
一、地中海贫血概述关于地中海贫血,首先我们来谈一谈它的一些概述方面。
地中海贫血是怎么命名的地中海贫血是一种严重威胁人类健康的致死致残的遗传性的血液疾病,它最早是由美国的儿科医生Tomas B.Cooley 于1925 在描述β地中海贫血的时候,首次报道的,所以被称为Cooley 贫血,这个是地中海贫血命名的一个原由。
但是由于早期报道的病例几乎都来自地中海地区的移民,因此我们把这种病命名为地中海贫血,简称地贫,后来证实其实地中海贫血在地中海沿岸国家以外的热带和亚热带地区,比如中国的南方,东南亚地区,中东地区都是高发地区,所以实际上,地中海贫血不仅仅存在与地中海沿岸国家,它还存在以外的一些热带和亚热带的地区。
我们可以看看,地中海贫血在全世界的一个分布的区域图,从这里我们看到,黄色区域就是地中海贫血的一个分布的发病区域,地中海贫血是全球最大的单基因遗传病之一,在我们中国不同的地区,特别是在南方的高发地区,它的人群携带率可以高达23% 左右。
在20 世纪60 年代,我们研究发现,地中海贫血是由α和β株蛋白比例不平衡,导致血红蛋白的合成缺陷,红细胞容易破裂,从而导致的一个溶血性的贫血,到20 世纪70 年代,我们先后发现了α、β株蛋白肽链的基因,它是基因组的形势存在的,α珠蛋白基因朱定义为16 号染色体的16p13.3 位点,而β珠蛋白基因族定位于11 号染色体的11p15.3 位点。
地中海贫血既然是一个最大的单基因遗传病,我们来看一看它的遗传规律,从这个幻灯我们可以看到,地中海贫血它是一种典型的常染色体的隐性遗传疾病,这就意味着如果母亲和服父亲都是隐性地贫基因的携带者,他们双方轻型的时候是没有临床表现的,和正常人是一样的,所以你看不出来他具有地中海贫血,但是如果他们两个结婚以后,在生育的后代,就会出现种情况,第一种是完全正常,第二种是基因携带者。