一例线粒体脑病患者的药例分析
- 格式:ppt
- 大小:861.00 KB
- 文档页数:20


2016.11护理经验233一例线粒体脑肌病的护理左丽平杭州市滨江医院神经内科 浙江省杭州市 310009【摘 要】目的:探讨一例线粒体脑肌病的护理以及经过护理的临床效果。
方法:2012-12-17我科收住一例,经过精心治疗和护理。
结果:该患者病情好转出院。
结论:根据患者的临床表现制定一个完整的护理计划,避免因不适当的护理或不适当的生活方式导致病情加重。
早期护理包括主要有视力、智力的减退,全身无力,运动不耐受,对其应该给予心理护理,多与患者沟通,协助患者完成日常生活,中晚期的护理重点是并发症上的护理,患者有可能出现全身肌肉萎缩,偶发癫痫,继尔可有多脏器功能的衰竭,出现呼吸困难等,这时的护理重点应该是应防止患者受伤、保证呼吸道的通畅、防止压疮的发生。
【关键词】线粒体脑肌病;护理;体会线粒体脑肌病是一组由于线粒体功能缺陷引起的多系统疾病,以中枢神经和肌肉系统病变为基础,特征是呼吸链酶活性正常的肌纤维与酶活性缺失的肌纤维混合,如病变同时累及到中枢神经系统,则称为线粒体脑肌病[1]。
根据临床特征可分为多个不同类型,常见的有以下几种:(1)伴有破碎红纤维的肌阵挛癫痫(MERRF )(2)线粒体脑肌病合并乳酸血症及卒中样发作(MEIAS ),慢性进行性眼外肌麻痹,Kearns —Sayre 综合征,卒中样发作是线粒体脑肌病一个重要的临床表现。
神经元兴奋性增高是疾病发展的关键环节,卒中样发作与癫痫 、偏头痛等发作性疾病存在着相似的电生理过程,也可以说在线粒体脑病中,癫痫,偏头痛本身就是一种卒中样发作形式[2]。
线粒体遗传病是近40多年来发现的一个新的疾病体系,2012-12-17我科收住一例,经过精心治疗和护理,结果患该者病情好转出院,就此病我的学习体会如下:1 资料与方法1.1 一般资料患者16-1沈晓康,青年男性,16岁,因“头痛头晕3天,视力下降1天”于2012-12-17入院。
主要表现为视力明显下降,头痛头晕,全身疲乏无力,查体:T:37.1℃,P:112次/分,R:18次/分,BP117/90mmHg ,精神欠佳,双眼视力下降,口齿清,四肢肌力V 级。

癫痫药物治疗研究进展魏惠;赵文艳;高学军【期刊名称】《临床误诊误治》【年(卷),期】2017(030)006【总页数】5页(P108-112)【关键词】癫痫;药物疗法;研究【作者】魏惠;赵文艳;高学军【作者单位】716000 陕西延安,延安大学附属医院心脑血管病医院神经内科;716000 陕西延安,延安大学附属医院心脑血管病医院神经内科;716000 陕西延安,延安大学附属医院心脑血管病医院神经内科【正文语种】中文【中图分类】R742.1据世界卫生组织统计,全球大约有5000万癫痫患者[1]。
国内流行病学资料显示,目前我国约有900万以上的癫痫患者,其中500万~600万为活动性癫痫患者,同时每年新发患病人数65万~70万[2]。
临床研究表明,新发癫痫患者若接受规范、合理的抗癫痫药物(antiepileptic drugs, AEDs)治疗,70%~80%的患者癫痫发作是可控制的,其中60%~70%的患者经2~5年的药物治疗可停药观察[3]。
因此,AEDs是治疗癫痫最重要、最基本的方法。
在AEDs治疗过程中,关注疗效的同时要观察药物不良反应,尤其是对特殊人群的不良影响。
本文就近年治疗癫痫的药物临床应用做一简要综述。
1.1 传统AEDs1.1.1 丙戊酸钠(valproate, VPA):为一种广谱AEDs,是全面性发作尤其是全面强直痉挛发作并典型失神发作的首选药物,也可用于部分性发作及双向情感障碍相关的躁狂发作的治疗。
主要通过抑制Na+通道和T-Ca2+通道,增加神经细胞突触后γ-氨基丁酸(GABA)亲和力,增强GABA通道功能以控制癫痫异常脑电波的发放和播散,同时还可抑制N-甲基-D-天门冬氨酸(NMDA)受体介导的神经兴奋从而发挥抗癫痫作用[4]。
VPA半衰期为8~15 h,血浆蛋白结合率为90%~95%,若同时服用其他蛋白结合率较高的药物(如阿司匹林),可增加VPA的血浆游离浓度,引发中毒。

伴小脑萎缩的线粒体脑肌病MELAS一例作者:张忠胜石喆来源:《新医学》2021年第07期【摘要】线粒体脑肌病伴高乳酸血症和卒中样发作(MELAS)是一种少见的遗传性疾病,可累及机体多系统。
该病主要临床表现为头痛、癫痫、耳聋、皮质盲及认知功能下降等。
MELAS呈卒中样发作,临床易误诊为脑梗死及脑炎,目前尚缺乏特效治疗方法。
该文报道1例33岁女性MELAS 患者,其以突发头痛、视物不清为首发症状,伴有不完全感觉性失语、听力下降、不能耐受疲劳,急诊颅脑CT显示双侧小脑半球萎缩,入院后经外周血基因检查明确MELAS诊断,予辅酶Q10胶囊、艾地苯醌及维生素E治疗。
患者病情好转后出院,随访3个月病情稳定。
该病例提示临床医师应提高对MELAS的认识,注意鉴别诊断,避免漏诊或误诊。
【关键词】线粒体脑肌病;小脑萎缩;卒中样发作;病例报告MELAS syndrome complicated with cerebellar atrophy: a case report Zhang Zhongsheng, Shi Zhe. Department of Neurology, the 6th Affiliated Hospital of Guangzhou Medical University (Qingyuan People’s Hospital), Qingyuan 511518, China Corresponding author, Shi Zhe, E-mail:155****************【Abstract】Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) is a rare hereditary disease, which can involve with multiple systems. The main clinical manifestations of MELAS consist of headache, epilepsy, deafness, cortical blindness and cognitive decline, etc. MELAS presents with a stroke-like episode, which is easily misdiagnosed as cerebral infarction and encephalitis. No specific treatment has been available at present. In this article, one 33-year-old female with MELAS was reported, presenting with sudden headache and blurred vision as the first symptoms, accompanied by incomplete sensory aphasia, hearing loss,and intolerance of fatigue, etc. Emergency brain CT scan showed bilateral cerebellar atrophy. The diagnosis of MELAS was confirmed by peripheral blood genetic examination. Coenzyme Q10 capsules, idebenone tablets and vitamin E were given. The patient was discharged after improvement of the disease and remained physically stable after 3 months of follow-up. This case prompts that doctors should deepen the understanding of MELAS, pay attention to differential diagnosis and avoid missed diagnosis or misdiagnosis.【Key words】Mitochondrial encephalomyopathy;Cerebellar atrophy;Stroke-like episode;Case report線粒体脑肌病是一种由线粒体DNA或核DNA缺陷导致线粒体结构和功能障碍,使肌纤维和脑神经细胞ATP生成不足而引起的肌肉和(或)脑的病变。

线粒体神经胃肠脑肌病一例临床、病理及基因分析2014-03-24 19:36来源:中华神经科杂志作者:唐吉刚等字体大小-|+线粒体神经胃肠脑肌病(MNGIE) 是一组以胃肠道症状、恶液质、周围神经病、眼外肌麻痹、白质脑病为临床表现的线粒体病。
目前国内有关该病的临床、病理及基因研究报道较少。
我们报道1 例临床表现典型的MNGIE 患者,对其临床、病理、线粒体及脱氧胸腺嘧啶核苷磷酸化酶(thymidinephosphorylase,TP) 基因进行分析。
临床资料患者男性,21 岁,因腹胀7 年、十二指肠空肠吻合术后1 个月于2012 年10 月8 13 收入我院普外科。
患者自13 岁左右经常出现腹胀、腹痛,间有腹泻症状,并逐渐出现消瘦,在当地医院诊断十二指肠淤滞症,于2012 年9 月8 日行十二指肠(水平段) 空肠吻合术。
术后12 d 患者出现腹胀、呕吐、腹泻,胃镜检查示反流性食管炎、胃潴留,遂转我院普外科。
图1 患者头颅MRI 示T2 液体衰减反转恢复序列成像可见双侧放射冠、侧脑室旁高信号2012 年10 月19 日无明显感染诱因出现双下肢无力,并逐渐加重,约20 d 后出现双上肢远端无力,肌电图检查示周围神经病变,脑脊液检查示脑脊液蛋白.细胞分离,经神经内科会诊考虑吉兰- 巴雷综合征,给予人免疫球蛋白(0.4 g·kg-1·d-1) 治疗,病情无明显变化。
11 月19 日行头颅MRI 检查示双侧放射冠、侧脑室旁、胼胝体、脑干、小脑半球多发等、长T2 异常信号(图1),遂转至我院神经内科继续诊治。
既往史:患者儿童期发育正常,因自幼被现在家庭抱养,家族遗传史无从查询。
入院体检:体温37℃,脉搏126 次/min,呼吸16 次/min,血压140/80 mmHg(1 mmHg=0.133kPa)。
体形消瘦,营养差,较标准体重减低约35%,心、肺体检无明显异常。
舟状腹,上腹部一长约15cm 手术瘢痕,腹部皮褶约0.4 cm,全腹无明显压痛、反跳痛,振水音阳性。

一例线粒体脑病患者的药例分析线粒体脑病(mitochondrial encephalopathy)是一种罕见的、遗传性的神经变性疾病,主要影响中枢神经系统和周围神经系统,严重影响患者的生活质量。
以下是一例线粒体脑病患者的药物治疗例分析。
患者信息:性别:男年龄:37岁初次就诊日期:2024年1月5日主述症状:患者主述持续性的头痛和乏力感,已经有两年的时间。
此外,他还表现出持久性的嗜睡和注意力不集中。
体检发现他有眼睑下垂、口吃、吞咽困难、四肢微弱无力等症状。
他的两个姐姐也有类似症状。
临床检查:神经系统检查发现患者有共济失调、震颤和麻木感。
CT和MRI扫描显示脑部有一定程度的萎缩。
线粒体功能测定结果显示乳酸和丙酮酸水平升高。
线粒体DNA测序结果发现线粒体DNA缺失。
根据上述临床表现和检查结果,确诊为线粒体脑病(mitochondrial encephalopathy)。
药物治疗方案:1. 辅酶Q10片(Coenzyme Q10 tablets):每天口服400mg,分为两次。
辅酶Q10是线粒体电子传递链中的一个重要成分,可提高线粒体功能。
2. 甲氰咪胍片(Metformin tablets):每天口服1000mg,分为两次。
甲氰咪胍作为一种抗糖尿病药物,能够提高线粒体功能和代谢。
3. 普鲁卡因胺注射液(Procainamide injection):每天静脉注射1000mg,分为三次。
普鲁卡因胺作为一种抗心律失常药物,可以改善心脏的线粒体功能,减轻心肌炎症。
4. 甲氨蝶呤片(Methotrexate tablets):每周口服15mg,分为三次。
甲氨蝶呤是一种免疫抑制剂,适用于治疗线粒体脑病的免疫反应。
5.肾上腺皮质激素:每天口服泼尼松或地塞米松,剂量根据病情调整。
肾上腺皮质激素可通过抑制免疫系统的活动来减轻炎症反应。
6.辅助治疗:包括镇静剂和抗抑郁药,以改善患者的睡眠和情绪状态。
治疗效果和观察指标:在第一次药物治疗后,患者的头痛和乏力感有所减轻,嗜睡和注意力不集中的症状有所改善。


乙基丙二酸脑病患儿的临床研究及ETHE1基因新突变1例报告李溪远1 丁圆1 刘玉鹏1 王峤1 宋金青1 叶锦棠2 张尧1 吴桐菲3 杨艳玲1【摘要】摘要:目的1例经EHTE1基因分析确诊的罕见的常染色体隐性遗传病——乙基丙二酸脑病。
方法回顾性分析1例确诊乙基丙二酸脑病患儿的临床经过、基因突变特点等。
结果女性患儿,7个月起出现顽固性腹泻,并逐渐出现智力运动发育落后及倒退,23个月时检测患儿血液丁酰肉碱4.48 mmol/L,异戊酰肉碱0.7 mmol/L;尿乙基丙二酸及甲基琥珀酸浓度显著增高;颅脑MRI显示双侧基底节区对称性异常信号,符合乙基丙二酸脑病;ETHE1基因分析发现2个杂合突变,其中c.488G>A(p.R163Q)为已知突变,c.203T>C(p.L68P)为未报道的新突变。
患儿经免乳糖饮食,以及左卡尼汀、辅酶Q10、维生素B1、维生素B2、维生素C支持治疗后,全身情况改善,智力运动发育有所进步,但仍明显落后。
结论尿液乙基丙二酸增高可见于线粒体脂肪酸代谢障碍等多种疾病,血液丁酰肉碱、异戊酰肉碱浓度增高是乙基丙二酸脑病的常见生化表现之一,ETHE1基因分析有助于确诊。
【期刊名称】临床儿科杂志【年(卷),期】2014(000)010【总页数】5【关键词】乙基丙二酸尿症;乙基丙二酸脑病; ETHE1基因;有机酸尿症;儿童乙基丙二酸脑病(ethylmalonic encephalopathy,OMIM #602473)是一种罕见的常染色体隐性遗传代谢病,1991年由Burlina等[1]首次报道。
乙基丙二酸脑病是由于ETHE1基因缺陷,而导致脑、胃肠、血管多脏器损害,以持续性乙基丙二酸尿症、智力运动落后、顽固性腹泻、瘀点、直立性手足发绀及大脑灰质损害为主要特征[2-4]。
乙基丙二酸脑病患儿临床表现相似,常于出生半岁以内发病,2岁以内因代谢代偿失调或脑功能衰竭死亡,病死率、残障率均很高[4,5]。
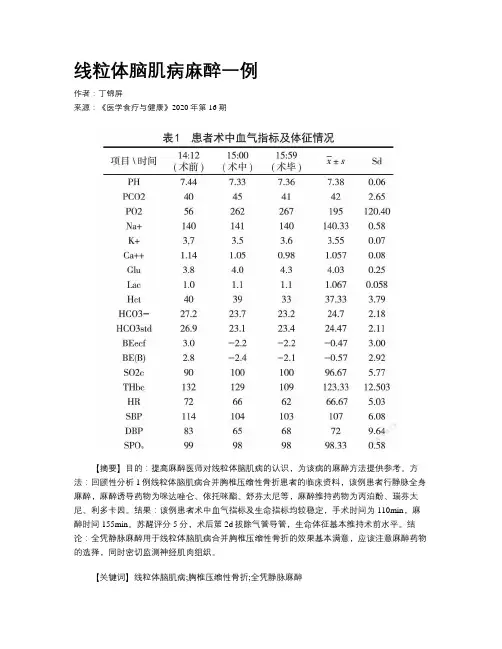
线粒体脑肌病麻醉一例作者:丁锦屏来源:《医学食疗与健康》2020年第16期【摘要】目的:提高麻醉医师对线粒体脑肌病的认识,为该病的麻醉方法提供参考。
方法:回顾性分析1例线粒体脑肌病合并胸椎压缩性骨折患者的临床资料,该例患者行静脉全身麻醉,麻醉诱导药物为咪达唑仑、依托咪酯、舒芬太尼等,麻醉维持药物为丙泊酚、瑞芬太尼、利多卡因。
结果:该例患者术中血气指标及生命指标均较稳定,手术时间为110min,麻醉时间155min。
苏醒评分5分,术后第2d拔除气管导管,生命体征基本维持术前水平。
结论:全凭静脉麻醉用于线粒体脑肌病合并胸椎压缩性骨折的效果基本满意,应该注意麻醉药物的选择,同时密切监测神经肌肉组织。
【关键词】线粒体脑肌病;胸椎压缩性骨折;全凭静脉麻醉[中图分类号]R746 [文献标识码]A [文章编号]2096-5249(2020)16-0-02[Abstract] Objective: To improve the understanding of anesthesiologists on mitochondrial encephalomyopathy and to provide a reference for anesthesia methods for the disease. Methods: The clinical data of a patient with mitochondrial encephalomyopathy and thoracic vertebral compression fracture were retrospectively analyzed. The patient underwent intravenous general anesthesia. The induction drugs were midazolam, etomidate, sufentanil, and anesthesia maintenance drugs Lidocaine, propofol, remifentanil. Results: The blood gas index and life index were stable during the operation, The operation time was 110 minutes and the anesthesia time was 155 minutes. The awake score was 5 points, and the tracheal tube was removed on the 2nd day after the operation,and the vital signs basically maintained the preoperative level. Conclusion: The effect of total intravenous anesthesia on mitochondrial encephalomyopathy combined with thoracic vertebral compression fractures is basically satisfactory. Attention should be paid to the selection of anesthetic drugs, while strengthening the monitoring of neuromuscular tissue during the operation.[Key words] Mitochondrial Encephalomyopathy; Thoracic Vertebral Compression Fracture; Total Intravenous Anesthesia线粒体是细胞进行有氧呼吸的主要场所,也是除nDNA外的遗传物质[1]。

儿童线粒体病6例临床分析陈秉洁蔡文红林云峰王世彪张宝泉福建省妇幼保健院福建医科大学附属医院新生儿科,福州350001通信作者:林云峰,Email:【摘要】目的探讨线粒体疾病临床特点及预后。
方法回顾本院2012年1月至2020年1月收治6例线粒体疾病患儿,包括询问病史、总结临床表现、实验室检查、影像学检查、基因检测结果及随访情况。
结果6例患儿中男3例,女3例;发病年龄范围为0~12.0岁,中位发病年龄4.09岁,新生儿期发病2例,1岁以内发病3例(50.0%);神经系统症状4例,伴有惊厥病史2例,生长发育落后3例;随访已死亡2例,病愈1例。
结论基因诊断可从婴幼儿严重线粒体疾病患者中区别出预后良好的患者,给予相应的支持治疗,帮助其度过危险时期。
【关键词】线粒体病;可逆性;儿童;新生线粒体病(MD)是一组由于线粒体DNA(mitochondrial DNA)或核DNA(nuclear DNA)缺陷导致线粒体结果和功能异常引起的一组疾病,临床上具有多样化表现,该病常同时累及多个器官,目前临床有多种综合征被证实属于线粒体疾病,近年来随着对线粒体病检测技术的提高,线粒体病的基因型、临床表型和诊断治疗迅速发展;澳大利亚的一项研究估计,16岁之前儿童线粒体疾病患病率为6.2/100000[1],可逆性婴幼儿呼吸链缺乏症(reversible infantile respiratory chain deficiency,RIRCD)罕见报道。
典型的临床表型包括:乳酸酸中毒及卒中样发作的线粒体脑肌病(MELAS),Leigh 综合征(LS)、Pearson综合征等,另外临床发现一类型MD为可逆性病变,新生儿或婴儿起病的由编码tRNAGlu的MT-TE基因m.14674T>T/C突变引起RIRCD[2],本文回顾分析2012年1月至2020年1月本院收治6例线粒体疾病患儿临床特点、实验室检查及基因型,以期为临床提供参考。

线粒体脑肌病伴高乳酸血症和卒中样发作1例陈旭辉;吴军;陈涓涓;林凯华【期刊名称】《罕少疾病杂志》【年(卷),期】2015(000)004【总页数】3页(P9-10,27)【关键词】线粒体脑肌病;卒中样发作;影像学;肌肉病理学【作者】陈旭辉;吴军;陈涓涓;林凯华【作者单位】北京大学深圳医院神经内科广东深圳 518000;北京大学深圳医院神经内科广东深圳 518000;北京大学深圳医院神经内科广东深圳 518000;北京大学深圳医院神经内科广东深圳 518000【正文语种】中文【中图分类】R651.1现病史:患者,女性,39岁,以“发作性四肢抽搐、神志不清2月,加重半天余”急诊入院。
患者2月前在睡眠中突发四肢抽搐,表现为四肢强直,头部转向右侧,双眼眼球上翻,呼吸急促,历时约5分钟后四肢抽搐缓解,2小时内共约发作10次,送至当地医院急诊科,予“地西泮、咪达唑仑、鲁米那”等治疗,并行气管插管、呼吸机辅助呼吸等治疗,查头颅CT提示“左侧基底节区软化灶”,查头颅MR发现颅内多发病灶,自身抗体提示多项抗体阳性,考虑诊断为:结缔组织病,出院后坚持口服“丙戊酸钠缓释片0.5 bid”。
4月13日患者在睡眠中再次反复出现四肢抽搐及意识障碍,同样予气管插管、呼吸机辅助呼吸、“丙戊酸钠、鲁米那”抗癫痫、“哌拉西林/他唑巴坦”抗感染治疗,复查头颅MR提示“双侧额顶叶病灶较前增多,左侧基底节区、左侧小脑半球病灶较前缩小”,并诊断为:系统性红斑狼疮?干燥综合征?出院后服药抗癫痫治疗。
昨天晚上23时许患者在睡眠中再次反复出现四肢抽搐伴意识障碍,尿失禁一次,6小时后送至我院急诊科,在急诊科就诊期间仍有发作,予“鲁米那”肌肉注射等治疗,症状未完全控制,拟“癫痫,结缔组织病”收入我科。
既往史:出生时曾经使用产钳、有脐带绕颈。
7岁左右出现癫痫大发作,每2~3个月发作一次,严重时每周发作三次,9岁开始口服中药治疗后未再发作,11岁左右停药后未再发作。
依达拉奉在神经科疾病中的应用瞿剑峰【摘要】依达拉奉是一种新型的自由基清除剂,其具有强大的清除自由基功能,能抑制脂质过氧化、减少氧化应激等.除了已广泛应用于缺血脑卒中的治疗外,近来还发现对出血性脑卒中、脑和脊髓损伤、神经退行性病变、癫痫、周围神经病以及神经痛等疾病也具有潜在的治疗作用.该文着重对依达拉奉在神经科各类疾病中的基础研究和临床应用进展进行归纳、整理和分析,为依达拉奉在神经科的临床应用提供科学依据.【期刊名称】《医学综述》【年(卷),期】2014(020)009【总页数】3页(P1641-1643)【关键词】依达拉奉;中枢神经;临床应用【作者】瞿剑峰【作者单位】上海市奉贤区中心医院神经内科,上海 201499【正文语种】中文【中图分类】R741.05依达拉奉(edaravone,MCI-186),化学名为3-甲基-1-苯基-2-吡唑啉-5-酮(3-Methyl-1-phenyl-2-pyrazolin-5-one),于2001年在日本上市,作为一种新型的自由基清除剂,能抑制脂氧合酶参与的花生四烯酸代谢,抑制四氧嘧啶诱导的脂质过氧化,刺激前列环素的生成,抑制炎性介质(如白三烯)形成,猝灭中性粒细胞产生H2O2和·OH[1]。
其在脑缺血的治疗中作用肯定[2],在其他的神经系统疾病中也有潜在的作用。
1 脑卒中1.1 缺血性脑卒中依达拉奉自上市以来已被广泛用于治疗缺血性脑卒中。
Feng等[3]对包括Cochrane(科克伦)脑卒中登记组和中国卒中试验登记组在内的三项已完成的试验和四项待评价的试验进行分析以明确依达拉奉对急性缺血性卒中的疗效和安全性,结果发现每日静脉滴注依达拉奉60 mg,共14 d,能明显促进患者神经功能改善,与对照组相比差异显著。
国外的实践也证明了依达拉奉安全有效[4]。
1.2 脑出血 Nakamura等[5]发现,在大鼠脑出血后立即或2 h时经全身给予依达拉奉能减少出血24 h后的脑含水量,减轻脑水肿,改善神经功能缺损;能减轻脑出血引起的嘌呤/脱嘧啶碱基位点和8-羟基-2′-脱氧鸟苷变化,减轻氧化损伤;降低铁和凝血酶诱导脑损伤。
1例Wernicke脑病患者的护理作者:冯晓瑜,曹迎春,刘捷来源:《护理实践与研究》 2016年第18期冯晓瑜,曹迎春,刘捷doi:10.3969/j.issn.1672-9676.2016.18.078Wernicke脑病由于维生素B1(即硫胺)缺乏引起的中枢神经系统的代谢性疾病,可导致脑组织对葡萄糖的利用率减小、脑细胞乳酸酸中毒、氧化应激、内皮细胞功能障碍及一氧化氮所致线粒体功能障碍等,最终导致神经细胞凋亡[1-2],主要表现为突然发作的神经系统功能障碍,典型的Wernicke脑病出现眼肌瘫痪、共济失调、精神及意识障碍三联征[3],临床上多数患者仅表现出三联征中的1~2项。
本病由Carl Wernicke于1881年首先报道,当时描述的3例患者为急性起病,均以死亡为结局,病理解剖后发现为血管损害,累及了脑室和灰质。
我院2015年11月收治1例Wernick脑病患者,现报道如下。
1病例介绍患者,青年女性,病程6个月余,亚急性起病。
半年前无明显诱因出现反复呕吐胃内容物1月余,当地医院就诊予以对症治疗后好转,4个月前患者开始出现天旋地转样头晕、阵发性头痛,伴有听力下降及高调耳鸣。
2个月前患者出现夜间睡眠不佳,伴有烦躁、乱语等精神和意识障碍。
1个月前患者逐渐出现双眼睑下垂、复视及言语不清,近1周患者出现站作者单位:510120广州市广州医科大学附属第一医院神经内科冯晓瑜:女,硕士研究生,主管护师立及行走不稳,为求进一步诊治于2015年11月5日来我院就诊。
查体:神志清楚,构音清晰,对答切题,双侧眼裂正常,双侧瞳孔等大等圆,直径约3.0 mm,对光反射灵敏,双侧眼睑下垂,左眼外展受限,额纹、左侧鼻唇沟变浅,示齿口角歪向右侧,双侧咽反射正常,四肢肌张力正常,明显躯干性共济失调,闭目难立征阳性,左侧中枢性面瘫,左侧肢体偏瘫,左侧肢体感觉过敏,左侧轻瘫试验阳性,指鼻试验、跟-膝-胫试验正常,闭目站立征阳性,双侧病理征阴性,脑膜刺激征阴性。
全国052 诊断一直在闪光路上的线粒体脑肌病--安金范红珍齐亚超发言汇总滑动查看发言部分摘录滑动查看全部【发言汇总】「何晓非遂宁市中心医院神内:青年女性,急性起病,以突发头痛,恶心呕吐及视物模糊为主要表现,头颅磁共振无明显影响学异常,但脑电图发现大量癫痫样放电。
说明定位在广泛皮层损害,定性考虑免疫性?诊断:自免脑?感觉这个患者是一个发作性疾病,而且症状存在刻板性。
应该复查一下免疫包括ANCA,甲功是有问题的,也提示可能和免疫有关。
而且又是一个发作性疾病,中途有好转,又反复复发。
」- - - - - - - - - - -- - - -「陈银娟龙岩市一神内:青年女性,急性起病,反复发作,主要表现为头痛,呕吐,视物模糊。
定位:皮层颅内痛敏结构定性:可逆性血管收缩综合征?偏头痛?癫痫?自免脑?」- - - - - - - - - - -- - - -「张靖言保定中西医结合医院内六:患者,青年女,急性起病,主要表现头痛、恶心及视物模糊,定位:全脑颅内痛敏结构,定性:可逆血管脑炎线粒体脑病静脉窦瘘?」- - - - - - - - - - -- - - -「曾伟河池市医院:青年女性,亚急性起病,主要为头痛恶心呕吐,伴有视物模糊,查体无阳性体征,腰穿问题不大,只能无异常,无精神异常,脑电图异常,考虑定位皮层,或者脑膜,可逆性血管收缩综合征?静脉问题?病毒相关性脑炎?有明显头痛,脑电图有很多棘慢波,说明皮层明显兴奋,还是应该考虑皮层病变,感染脑脊液明显不支持,自身免疫性脑炎病灶好像不符合,没有强化,脑电图呈弥漫性,也不像,病毒相关性脑炎?」- - - - - - - - - - -- - - -「张乐国沧州市中心医院神内二:目前先猜个短暂头痛、神经功能缺损伴脑脊液淋巴细胞增多综合征(HaNDL syndrome)。
待继续放水。
」- - - - - - - - - - -- - - -「陈升东~102医院神内:青年女性,以头痛恶心呕吐为主,伴有视物模糊,查体无阳性体征,检查仅脑电图异常,核磁异常,考虑定位皮层,颅内痛敏结构,大脑后动脉有问题,可逆性血管收缩综合征?脱髓鞘?自免脑?」- - - - - - - - - - -- - - -「苏曼北京大学深圳医院:青年女性,反复发作性程度较剧烈头痛,视幻觉,脑电图异常电活动,持续一段时间后逐渐缓解,头部MRI提示有多部位的病灶,定位颅内多发病变,病变较为广泛,脑电图可逆性异常,定向:血管性:可逆性脑血管收缩?,血管炎性?;能量代谢性:线粒体脑病?」- - - - - - - - - - -- - - -「韩凌菏泽市立医院神经内:青年人,急性起病,表现头痛,恶心,视物模糊,高颅压症状,定位,全脑,定性,考虑脑炎?静脉窦?瘘?」- - - - - - - - - - -- - - -「张志刚商河中医院内科:定位,广泛大脑皮层,定性,考虑感染」- - - - - - - - - - -- - - -「张燕定州市中医医院神内:青年女性,急性起病,反复发作,主要表现为头痛,呕吐,视物模糊。
线粒体病4例的临床分析目的探讨线粒体病的临床特征及诊断。
方法回顾性分析4例线粒体病患者的临床特征和辅助检查结果。
结果4例患者共同症状为肌肉耐疲劳性差,主诉易感疲劳,其余症状表现多样,运动乳酸试验均阳性,肌肉活检最后确诊。
结论线粒体病临床不多见,临床症状不典型,易误诊漏诊,运动乳酸试验是协助早期鉴别诊断线粒体病的有效、简单的方法。
标签:线粒体病线粒体脑肌病运动乳酸试验线粒体病(Mitochondrial disorders)是遗传缺陷损伤引起线粒体代谢酶缺陷,使ATP合成障碍,能量来源不足导致的一组异质性病变。
现将我院收治的4例确诊病例回顾分析。
1、临床资料1.1一般资料:2011年至今收治4例线粒体病患者。
其中男性3人,女性1人。
年龄37-51岁。
女性病人妹妹有类似病史,27岁死于支气管扩张并感染,母亲及另一姐姐正常。
1.2 主要症状及体征:本组中全部病人均有肢体乏力症状,主诉易疲劳。
3例有视力下降,其中继发性青光眼及虹膜睫状体炎1例,双眼视神经萎缩1例,眼底检查正常1例。
肢体震颤1例,行走不稳1例,肌萎缩2例,弓形足1例。
肌力正常2例,肌力稍差2例。
腱反射表现各不相同,难立征加强试验(+)1例。
全部患者深浅感觉均正常。
病理征阳性1例。
1.3 辅助检查:4例患者均进行三大常规、肝肾功能、凝血功能、血糖、血脂、甲状腺功能、性激素六项、多肿瘤标志物、风湿、免疫指标、ANA、抗ENA、抗ds-DNA检查,结果正常。
2例进行腰穿术检查颅内压正常,脑脊液生化及常规正常。
1例患者CK升高。
肌电图表现为全身广泛性神经源性损害1例,表现为四肢周围神经受累1例,表现为肢体感觉纤维重度混合性损害、远端运动纤维轻至中度轴索损害,神经源性损害1例,肌电图正常1例。
脑电图异常1例。
头颅MR全部未见异常。
有肌萎缩表现的2例患者中1例胸椎MR增强提示胸1~2节段脊髓片状异常信号,无强化,无占位效应,另1例颈、胸椎MR未见异常。
线粒体MT-TE基因突变致可逆性婴幼儿呼吸链缺乏症1例并文献复习罗序峰;刘楠;谢惠源;朱建萍;付四毛;周涛;张胜【摘要】目的探讨线粒体MT-TE基因突变致可逆性婴幼儿呼吸链缺乏症的临床表现及基因突变特点.方法回顾分析1例确诊为可逆性婴幼儿呼吸链缺乏症患儿的临床资料,并复习相关文献.结果男性患儿,2个月23天,入院时消瘦,呼吸急促,双肺可闻及痰鸣音及喘鸣音;肌力IV级,肌张力减弱.有一胞姐生后不久因重症肺炎去世.入院时肌酸激酶同工酶123 U/L,肌酸激酶890 U/L;血乳酸8.9 mmol/L;病原学检查均为阴性.头颅MRI无异常.入院后患儿持续高乳酸血症、肌酶异常升高,伴呼吸困难,放弃治疗后死亡.基因检查示线粒体MT-TE基因存在14674 T>C突变,来源于母亲.国外文献报道线粒体MT-TE 14674T>C突变患儿早期临床表现与进展型线粒体病类似,呼吸肌无力、喂养困难,运动发育里程碑延迟,肌酶及乳酸升高.予补充能量,维持内环境稳定等治疗,约1岁左右逐渐好转.结论线粒体MT-TE基因突变致可逆性婴幼儿呼吸链缺乏症早期表现与进展型线粒体病类似,积极治疗预后良好.早期进行基因检测可明确预后,增强治疗的信心.【期刊名称】《临床儿科杂志》【年(卷),期】2019(037)002【总页数】4页(P93-96)【关键词】可逆性;呼吸链缺乏症;MT-TE基因;线粒体肌病【作者】罗序峰;刘楠;谢惠源;朱建萍;付四毛;周涛;张胜【作者单位】深圳儿童医院工作;南方医科大学附属中山市博爱医院广东中山528400;南方医科大学附属中山市博爱医院广东中山 528400;南方医科大学附属中山市博爱医院广东中山 528400;南方医科大学附属中山市博爱医院广东中山528400;南方医科大学附属中山市博爱医院广东中山 528400;南方医科大学附属中山市博爱医院广东中山 528400【正文语种】中文线粒体病(mitochondrial diseases,MD)是由线粒体DNA(mtDNA)或核DNA(nDNA)突变引起线粒体代谢酶功能缺陷,导致三磷酸腺苷(ATP)合成障碍、能量产生不足而出现的一组多系统疾病。