变性梯凝胶电泳
- 格式:pptx
- 大小:1.59 MB
- 文档页数:21

sds-page凝胶电泳原理SDS-凝胶电泳(Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis)是一种常用的蛋白质分析和分离技术。
它基于蛋白质在电场中的迁移速率与分子质量之间的关系,通过将蛋白质样品加入SDS变性和还原缓冲液,并进行蛋白质的电泳分离,可以实现蛋白质分子量的估计和蛋白质组分的分离。
以下将详细介绍SDS-凝胶电泳的原理。
一、样品处理:在进行SDS-凝胶电泳之前,需要对样品进行处理,包括蛋白质的变性和还原。
这样做的目的是将蛋白质分子展开成线性构象,使各种蛋白质以相似的形式进入凝胶,并消除了蛋白质修饰对迁移速率的影响。
1.变性:加入SDS变性缓冲液。
SDS是一种带负电荷的表面活性剂,可以与蛋白质相互作用,让蛋白质线性展开,并赋予蛋白质负电荷。
在变性缓冲液中通常还会包含甘油和十二烷基硫酸钠,以提高蛋白质的溶解度。
2. 还原:加入还原剂。
通常使用二巯基乙醇(2-mercaptoethanol)或二巯基丙烷(Dithiothreitol, DTT)等还原剂,以破坏蛋白质的二硫键,消除蛋白质中的多肽链的二级结构。
二、凝胶制备:SDS-的凝胶通常由两种不同浓度的聚丙烯酰胺(polyacrylamide)单体制备而成,其中较稀的为分离凝胶,较浓的为聚合凝胶。
聚丙烯酰胺是由甲基丙烯酰胺(acrylamide)和交联剂(常用的是二甲基丙烯酰胺)共聚合而成的。
1. 分离凝胶(stacking gel):通常为4-15%(w/v)的聚丙烯酰胺。
分离凝胶中加入的缓冲液pH稍微较低,以产生较高的电场强度,使蛋白质迅速进入聚合凝胶。
2. 聚合凝胶(resolving gel):通常为8-20%(w/v)的聚丙烯酰胺。
聚合凝胶中加入的缓冲液pH较高,以产生较低的电场强度,使蛋白质在其中进行分离。
三、电泳操作:1.样品加载:将处理后的蛋白质样品加载到凝胶孔上。

6 %变性聚丙烯酰胺凝胶电泳实验试剂及配制:a, 40 %丙烯酰胺单体凝胶贮存液。
丙烯酰胺38 g,甲叉双丙烯酰胺 2 g,加双蒸水定容至 100mL,过滤,贮存于棕色瓶中。
b,变性剂。
1 M NaOH 1 mL,甲酰胺 95 mL,溴酚蓝 0. 05 g,二甲苯青 0. 05 g,加双蒸水定容至100 mL。
C, 固定液。
100 mL 冰醋酸溶于双蒸水定容至1 000 mL。
d, 银染液。
硝酸银 1 g, 37 甲醛 1. 5 mL,加双蒸水定容至1000 mL。
f, 显影液。
无水碳酸钠 30 g, 37 甲醛 1. 5mL, 10 mg /mL 硫代硫酸钠 200 μL,加双蒸水定容至 1 000 mL。
DNA 变性聚丙烯酰胺凝胶电泳的操作流程6 %变性凝胶的制备:40 %的凝胶贮存液 11. 2 mL,尿素36.36g, 5 × TBE 15mL,加双蒸水定容至 75 mL。
预冷至 4 ℃后,加入500 μL 10 过硫酸铵和 50 μL TEMD 混合均匀,灌胶。
灌胶完毕,插入样品梳,在室温下聚合 30 ~60 min。
聚合完全后,梳齿下可见二条折光线。
电泳:安装电泳装置,在电泳槽中加入1 × TBE缓冲液,使缓冲液覆盖样品孔。
拔出样品梳,用移液器吸取适量缓冲液冲洗样品孔。
打开变压器,恒定功率 60 W 预电泳 15 ~30 min。
预电泳结束后,用移液器将 DNA 样品小心注入样品孔中( 加样量为 3 ~ 5 μL) ,电泳直至带型分开。
固定:关闭电源,卸下玻璃板,剥离玻璃板,将胶板置于固定液中固定 30 min,直到指示剂颜色褪去。
漂洗:将胶板转移到双蒸水中漂洗三遍,每次 2 ~ 3 min。
染色:转移胶板至染色液中,在摇床上摇动染色 30 ~ 40 min。
显影:将胶板在双蒸水中漂洗 5 ~ 10 s 后立即放入预冷为 4 ℃~ 10 ℃的显影液中,摇动显影直到带型完全出现。

变性梯度凝胶电泳《变性梯度凝胶电泳》是一种有效的分离技术,它可以帮助科学家快速有效地分离不同类型的分子。
它和其他分离技术相比,有着独特的优势和不可替代的优势。
变性梯度凝胶电泳(DGGE)采用特殊的凝胶在不同变性环境中电泳,以有效地分离不同类型的DNA片段和蛋白质。
正常情况下,由于更多的低能量的片段的存在,特定的变性环境会吸引这些较低能量的片段,并且它们走的路程也会比较短,而其他高能量的片段则会走的路程会更长。
因此,将凝胶改变到一种低能量的片段可以吸引,而走得路程更长的片段,则可以有效地将这些片段分离开来。
DGGE的优点是它可以有效分离不同片段的DNA和蛋白质,并且可以更快地分离出更多的细小片段。
此外,由于DGGE有一个独特的低能量环境,它可以更好的区分不同的片段,而不会受到其他分离技术的限制。
另外,DGGE是一种半定量的分离技术,可以让研究者比较不同片段之间的相对含量。
除了上述优势,DGGE还具有一些缺点。
首先,由于梯度凝胶电泳系统搭建起来比较复杂,使用它来进行分离技术需要比较复杂的实验技术。
其次,由于梯度凝胶电泳系统比较复杂,对系统的调整和控制非常复杂,只有熟练的实验室人员才能处理好这些技术。
变性梯度凝胶电泳的应用非常广泛,它可以用来分离和分析不同类型的DNA片段和蛋白质,甚至可以用来检测细菌的抗药性。
它也可以用来研究基因的多样性,用于癌症和免疫系统的研究,以及对特定疾病的诊断。
最后,它也可以作为一种植物鉴定技术,用于比较不同植物类型间的分子差异。
总之,变性梯度凝胶电泳(DGGE)是一种有效的分离技术,它可以用于研究不同类型的DNA片段和蛋白质,还可以用于分析肿瘤细胞的分子结构,对特定疾病的诊断和植物鉴定。
它具有独特的低能量环境,可以更好的区分不同的片段,而不会受到其他分离技术的限制。
它具有快速、高效和半定量的优势,是研究DNA分子和蛋白质的有效分离技术。

变性梯度凝胶电泳摘要:变性梯度凝胶电泳(DenaturingGradientGelE1ectrophoresiS,DGGE)是由Fisher 和Lerman发明用于检测DNA突变的技术,其主要是基于突变型和野生型棱酸序列的不同而导致其变性浓度的差异,利用变性梯度凝胶进行分离, 该手段分辨率达一个碱基。
恒定变性凝胶电泳(CDGE)、瞬时温度梯度电泳(TTGE)和温度梯度凝胶电泳(TGGE)是由DGGE经改进而成。
它们在突变分析上都有着重要的作用。
关键词:变性梯度凝胶电泳、恒定变性凝胶电泳、瞬时温度梯度电泳、温度梯度凝胶电泳1、前言:随着结构基因组计划接近尾声,人类基因神秘的面纱已逐步揭开,GenBank中已知基因的数目也与日俱增,各种遗传病的相关基因也了解得越来越多。
但是,在找到了候选基因后,还需要在相关的遗传病病人中进行突变检测,只有找到了相应的突变。
才能真正确定该基因与疾病的关系,从而服务于临床。
变性梯度凝胶电泳就是一种很有实用价值的突变分析方法。
该方法经改进,又发展出了恒定变性凝胶电泳(constant denaturant gel elec—trophoresis,CDGE)、瞬时温度梯度电泳(temporaltemperature gradient electrophoresis,TTGE)和温度梯度凝胶电泳(temperature gradient gel elec—trophoresis,TGGE)。
现在上述方法已经广泛应用于遗传病的诊断、突变分析和肿瘤相关基因的筛查等许多方面。
2、基本原理2.1变性梯度凝胶电泳由Fisher和Izrman口3于1983年创立,以后该技术和PCR技术相结合被广泛应用于各种突变分析。
它主要是利用梯度变性胶来分离DNA片段。
电泳开始时,DNA在胶中的迁移速率仅与分子太小有关,而一旦DNA泳动到某一点时,即到达该DNA变性浓度位置时,使得DNA双链开始分开,从而大大降低了迁移速率。

变性梯度凝胶电泳(DGGE)和温度梯度凝胶电泳(TGGE)变性梯度凝胶电泳(DGGE)产品说明DGGE变性梯度凝胶电泳系统包括内置式温度控制系统、可编程控电源、梯度凝胶生成器、内置式缓冲液循环泵及搅拌桨、电泳槽、梯度生成器、制胶配件及玻璃板等。
DGGE介绍:变性梯度凝胶电泳法(denaturing gradient gel electrophoresis,DGGE)分析PCR产物,如果突变发生在最先解链的DNA区域,检出率可达100%,检测片段可达1kb,最适围为100bp-500bp。
基本原理基于当双链DNA 在变性梯度凝胶中进行到与DNA变性温度一致的凝胶位置时,DNA发生部分解链,电泳迁移率下降,当解链的DNA链中有一个碱基改变时,会在不同的时间发生解链,因影响电泳速度变化的程度而被分离。
由于变性梯度凝胶电泳法是利用温度和梯度凝胶迁移率来检测,需要一套专用的电泳装置,合成的PCR引物最好在5`末端加一段40bp-50bp的GC夹,以利于检测发生于高熔点区的突变。
DGGE原理:变性梯度凝胶电泳系统(denaturing gradient gel electrophoresis, DGGE)的原理是使用一对特异性引物,PCR扩增微生物自然群体的16s rRNA基因,产生长度相同但序列有异的DNA片段的混合物。
然后用变性梯度凝胶电泳分离产物混合物。
变性梯度凝胶电泳DGGE胶是在6%聚丙烯酰胺胶中添加线性梯度的变性剂,变性剂的浓度由上到下,从低到高成线性梯度。
在一定温度下,在同一浓度的变性剂浓度下,序列不同的产物,其部分解链程度也不同,而产物解链程度又直接影响其电泳迁移率,结果不同的产物在凝胶上分离开来。
在引物的5’端加上40个碱基左右的G-C串可使变性梯度凝胶电泳DGGE对序列差异的分辨率提高到近100%。
所以变性梯度凝胶电泳法可用于微生物群落结构的研究、微生物种群动态的分析、富集培养物及分离物的分析、核糖体RNA同源性的分析。
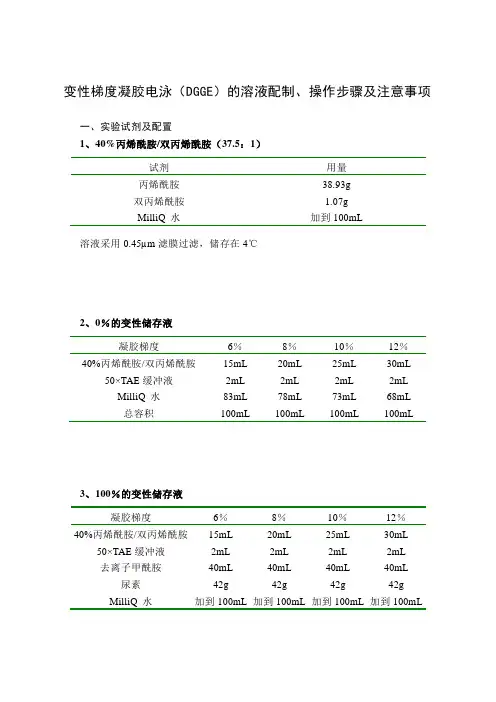
变性梯度凝胶电泳(DGGE)的溶液配制、操作步骤及注意事项一、实验试剂及配置1、40%丙烯酰胺/双丙烯酰胺(37.5:1)试剂用量丙烯酰胺38.93g双丙烯酰胺 1.07gMilliQ 水加到100mL 溶液采用0.45µm滤膜过滤,储存在4℃2、0%的变性储存液凝胶梯度6%8%10%12%40%丙烯酰胺/双丙烯酰胺15mL 20mL 25mL 30mL 50×TAE缓冲液2mL 2mL 2mL 2mL MilliQ 水83mL 78mL 73mL 68mL总容积100mL 100mL 100mL 100mL3、100%的变性储存液凝胶梯度6%8%10%12%40%丙烯酰胺/双丙烯酰胺15mL 20mL 25mL 30mL 50×TAE缓冲液2mL 2mL 2mL 2mL去离子甲酰胺40mL 40mL 40mL 40mL 尿素42g 42g 42g 42g MilliQ 水加到100mL 加到100mL 加到100mL 加到100mL4、50×TAE缓冲液试剂用量终浓度Tris碱242.0g 2M冰乙酸57.1 1M 0.5M EDTA,pH8.0 100.0mL 50mM MilliQ 水加到1000.0mL将溶液混合溶解,121℃下蒸汽灭菌20-30min,储存在室温5、银染液的配制:原液:8×固定液(250 ml):乙醇200 ml冰乙酸10mlMilliQ 水40ml(A):1×固定液(400 ml):8×固定液50mlMilliQ 水350ml(B)银染溶液(400ml):AgNO3 0.8 g8×固定液50mlMilliQ 水350ml(C):显影剂(500ml):NaOH 7.5gMilliQ 水500 ml甲醛 1.5ml6、10%过硫酸铵(AP):过硫酸胺0.3g超纯水 3.0ml溶解后使用0.45µm微孔滤膜过滤,4℃保存,保存时间为1周。


PCR-DGGE技术一、实验原理变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)最早是Lerman 等人于20 世纪80 年代初期发明的,起初主要用来检测DNA 片段中的点突变。
Muyzer 等人在1993 年首次将其应用于微生物群落结构研究。
后来又发展出其衍生技术,温度梯度凝胶电泳(TGGE)。
该技术被广泛用于微生物分子生态学研究的各个领域,目前已经发展成为研究微生物群落结构的主要分子生物学方法之一。
双链DNA分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为取决于其分子大小和电荷。
不同长度的DNA片段能够被区分开,但同样长度的DNA片段在胶中的迁移行为一样,因此不能被区分。
DGGE技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度,从而能够把同样长度但序列不同的DNA片段区分开来。
一个特定的DNA片段有其特有的序列组成,其序列组成决定了其解链区域和解链行为。
一个几百个碱基对的DNA片段一般有几个解链区域,每个解链区域有一段连续的碱基组成。
当变性剂浓度逐渐增加达到其最低的解链区浓度时,该区域这一段连续的碱基对发生解链。
当变性剂那浓度再升高依次达到其他解链区域浓度后,最高的解链区域也发生解链,从而双链DNA完全解链。
不同的双链DNA片段因为其序列组成不一样,所以其解链区域和解链区域的解链浓度也是不一样的。
当进行DGGE电泳时,一开始变性剂浓度比较小,不能使双链DNA片段最低的解链区域解链,此时DNA片段的迁移行为和在一般的聚丙烯酰胺凝胶中一样。
然而一旦DNA片段迁移到一特定位置,其变性剂浓度刚好能使双链DNA片段最低的解链区域解链时,双链DNA片段最低的解链区域立即发生解链。
部分解链的DNA片段在胶中的迁移速率会急剧下降。
因此同样长度但序列不同的DNA片段会在胶中不同位置处达到各自最低解链区域的解链变性剂浓度,因此它们会在胶中不同的位置分开。

RNA甲醛变性胶电泳提取样品的总RNA后,一般根据RNA的凝胶电泳图来判断RNA的质量。
由于RNA容易形成二级结构,因此常用甲醛变性胶来进行RNA电泳,得到的电泳图能真实反映RNA的质量状况。
一、试剂:DEPC(Sigma公司产品),MOPs(Bocherigmer公司产品),甲酰胺(Formamide,Sigma公司产品),溴酚蓝(Bromphenol Blue,Sigma公司产品)。
EDTA-Na2,氢氧化钠,醋酸钠,甲醛,甘油,分析纯,国产。
二、试剂配制:1、DEPC 水的处理:用量筒量取去离子水2L,在通风橱中,加入2ml DEPC到2升去离子水中,终浓度为0.1%的DEPC。
迅速盖上盖子,混匀,然后放在摇床中中速摇荡至少4hr,再高压灭菌。
灭菌时将瓶盖松开,15磅灭菌20min。
2、10 × FA Buffer(formaldehyde agarose buffer:200 mM的MOPs,50 mM的NaAc,10 mM的EDTA):配制500ml,称取3.4g NaAC•3H2O放入1000ml烧杯中,加入400ml DEPC 处理过的去离子水,加入搅拌子,放在磁力搅拌器上溶解。
然后加入20.9g MOPs,放在磁力搅拌器上溶解。
再加1.86g EDTA二水二钠,放在磁力搅拌器上溶解。
用1M灭过菌的NaOH 调PH至7.0(约用NaOH 40ml),用DEPC 处理过的去离子水定容至500ml,装入棕色瓶中,写好标签,室温避光保存。
4、5 ×甲醛变性胶加样缓冲液(5×loading buffer):配制10ml。
预先配制水饱和的溴酚蓝溶液:在1.5ml 离心管中加入约0.1mg溴酚蓝,加入1ml DEPC水溶解,充分振荡溶解,离心,可见离心管底部有溴酚蓝粉末剩余,上层液体即水饱和的溴酚蓝液。
在15ml灭菌离心管中,依次加入以下各种成分:4.0ml 10 × FA buffer3.1ml 甲酰胺2.0ml 100%的甘油720ul 37%的甲醛80ul0.5M的 EDTA (PH 8.0)16ul 水饱和的溴酚蓝100ul DEPC水混匀,分装,-20℃保存,常用放4℃保存。

RNA甲醛变性凝胶电泳
a、电泳槽用酒精棉球擦净,然后用3%H2O2浸泡10min,DEPC处理的水冲洗后封存80°C烘干;实验室用的所有玻璃器皿都要需要用0.1% DEPC处理的水溶液浸泡,37℃放置2h,然后用灭菌水淋洗数次,并于100℃干烤15min,在1.034×105Pa的压力下灭菌15min。
能经受高温烘烤的玻璃器皿可在洗净后,在150℃以上烤5h,以除去RNA酶。
b、铺胶(50ml)
琼脂糖0.75g(2.2%)
无RNase水36ml
10×MOPS 5ml
37%甲醛9ml
将琼脂糖加水用微波炉融化后,加10×MOPS,待胶冷却至60°C左右,再加甲醛和EB。
凝胶可以室温下放置1h;
c、RNA样品处理(以一条泳道为例)
10×MOPS 1.0ml
37%甲醛 3.5ml
甲酰胺10.0ml
RNA样品 4.5ml(20mg RNA)
EB 1ml
于65°C变性15min,冰浴冷却。
经此处理的RNA样品,-70°C可存放半年;
d、加4.0ml甲醛凝胶加样缓冲液(5×);
e、加样和电泳
将凝胶预电泳10min,电压为80v。
随后加样品,以电压为80v电压进行电泳,电泳液为1×MOPS 电泳缓冲液。
直至溴酚蓝迁移至胶下游的3/4处;
f、电泳结束后,取出凝胶,用DEPC处理过的水淋洗凝胶,然后进行RNA转膜;。

变性梯度凝胶电泳DGGE刘琳 1131428 环境科学一、 实验目的1. 学习掌握变性梯度凝胶电泳的原理和方法。
2. 练习变性梯度凝胶电泳的操作步骤。
3. 分析并掌握变性梯度凝胶电泳的思路,并了解其在微生物群落研究中的地位。
二、 实验原理双链DNA 分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为决定于其分子大小和电荷。
不同长度的DNA 片段能够被区分开,但同样长度的DNA 片段在凝胶中的迁移行为一样,因此不能被区分。
DGGE 技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度,从而能够把同样长度但序列不同的DNA 片段区分开来。
不同的双链DNA 片段因为其序列组成不一样,所以其解链区域及各解链区域的解链温度也是不一样的。
同样长度但序列不同的DNA 片段会在胶中不同位置处达到各自最低解链区域的解链温度,因此它们会在胶中的不同位置处发生部分解链导致迁移速率大大下降,从而在胶中被区分开来。
然而,一旦温度(或变性剂浓度)达到DNA 片段最高的解链区域温度时,DNA 片段会完全解链,成为单链DNA分子,此时它们又能在胶中继续迁移。
因此,如果不同DNA 片段的序列差异发生在最高的解链区域时,这些片段就不能被区分开来。
DNA 片段在DGGE 胶中的迁移行为(如下图):变性剂LowHigh在DNA 片段的一端加入一段富含GC 的DNA 片段(一般30-50个碱基对),使DNA 片段最高的解链区域在GC 夹子这一段序列处,它的解链温度很高,可以防止DNA 片段在DGGE/TGGE 胶中完全解链。
当加了GC 夹子后,DNA 片段中基本上每个碱基处的序列差异都能被区分开(Myers, 1985)。
DGGE 有两种电泳形式:垂直电泳和水平电泳。
垂直电泳是为了确定变性剂梯度;而水平电泳则是对比分析不同样品的微生物差异。
本次试验做的是水平电泳,主要对比分析不同样品的微生物差异。
三、 实验器材与药剂器材:DGGE system (D-Code, Bio-Rad)、移液管、移液枪、枪头、制胶设备、30ml 针筒、聚乙烯细管、电泳仪试剂:16s rDNA V3区PCR 扩增产物、胶浓度为8%变性剂浓度分别为0%和90%丙烯酰胺胶、50×TAE buffer、去离子甲酰胺、尿素、去离子水、10%过硫酸铵(Aps)、TEMED 、sDNA 染料 变性剂Low high四、实验步骤1.首先按照实验要求将50x TAE buffer稀释为1×TAE buffer,并利用90%和0%的变性胶分别配置15.5ml的35%和55%的变性胶溶液。
PCR-DGGE技术一、实验原理变性梯度凝胶电泳(denatured gradient gel electrophoresis,DGGE)最早是Lerman 等人于20 世纪80 年代初期发明的,起初主要用来检测DNA 片段中的点突变。
Muyzer 等人在1993 年首次将其应用于微生物群落结构研究。
后来又发展出其衍生技术,温度梯度凝胶电泳(TGGE)。
该技术被广泛用于微生物分子生态学研究的各个领域,目前已经发展成为研究微生物群落结构的主要分子生物学方法之一。
双链DNA分子在一般的聚丙烯酰胺凝胶电泳时,其迁移行为取决于其分子大小和电荷。
不同长度的DNA片段能够被区分开,但同样长度的DNA片段在胶中的迁移行为一样,因此不能被区分。
DGGE技术在一般的聚丙烯酰胺凝胶基础上,加入了变性剂(尿素和甲酰胺)梯度,从而能够把同样长度但序列不同的DNA片段区分开来。
一个特定的DNA片段有其特有的序列组成,其序列组成决定了其解链区域和解链行为。
一个几百个碱基对的DNA片段一般有几个解链区域,每个解链区域有一段连续的碱基组成。
当变性剂浓度逐渐增加达到其最低的解链区浓度时,该区域这一段连续的碱基对发生解链。
当变性剂那浓度再升高依次达到其他解链区域浓度后,最高的解链区域也发生解链,从而双链DNA完全解链。
不同的双链DNA片段因为其序列组成不一样,所以其解链区域和解链区域的解链浓度也是不一样的。
当进行DGGE电泳时,一开始变性剂浓度比较小,不能使双链DNA 片段最低的解链区域解链,此时DNA片段的迁移行为和在一般的聚丙烯酰胺凝胶中一样。
然而一旦DNA片段迁移到一特定位置,其变性剂浓度刚好能使双链DNA片段最低的解链区域解链时,双链DNA片段最低的解链区域立即发生解链。
部分解链的DNA 片段在胶中的迁移速率会急剧下降。
因此同样长度但序列不同的DNA片段会在胶中不同位置处达到各自最低解链区域的解链变性剂浓度,因此它们会在胶中不同的位置分开。
RNA的变性琼脂糖凝胶电泳实验原理、材料和操作步骤一、实验目的学习RNA琼脂糖凝胶电泳的使用技术。
二、实验原理RNA可以使用非变性或变性凝胶电泳进行检测。
在非变性电泳中,可以分离混合物中不同分子量的RNA分子,凡是无法确定分子量。
只有在变性情况下,RNA 分子完全伸展,其泳动率才与分子量成正比。
判断RNA提取物的完整性是进行电泳的主要目的之一。
完整的未降解的RNA 制品的电泳图谱应可清晰看到18s rRNA、28s rRNA、5s rRNA的三条带,且28s rRNA的亮度应为18s rRNA的两倍。
三、材料、试剂及器具1、材料总RNA提取物。
2、试剂(1)0.1%DEPC水:200ml双蒸去离子水加0.2mlDEPC焦炭酸、二乙酯混匀,室温放置过夜,高压灭菌。
(2)10x电泳缓冲液。
吗啉代丙烷磺酸(MOPS)0.4mol/L(pH 7.0)NaAc 0.1mol/L乙二胺四乙酸(EDTA) 10mmol/L(3)50mL变性琼脂糖凝胶(1%)(4)10x电泳缓冲液5 mL琼脂糖0.5 g0.1%DEPC水36.5 mL加热溶解,稍冷却,加入8.5 mL 37%甲醛。
(5)上样缓冲液:50%甘油,1mmmol/LEDTA,0.4% 溴酚兰,0.4%二甲苯蓝。
(6)甲酰胺(去离子)。
3、器具(1)电泳系统(2)紫外透视仪四、操作步骤1、将制胶用具用70%乙醇冲洗一遍,晾干备用。
2、制胶:称取0.5g琼脂糖粉末,加入放有36.5mL的DEPC水的锥形瓶中,加热使琼脂糖完全溶解。
稍冷却后加入5mL的10x电泳缓冲液、8.5mL的甲醛。
然后在胶槽中灌制凝胶,插好梳子,水平放置待凝固后使用。
3、加样:在一个洁净的小离心管中混合以下试剂:电泳缓冲液(10x)2μl、甲醛3.5mL、甲酰胺10mL、RNA样品3.5μl。
混匀,置60℃保温10min,冰上速冷。
加入3μl的上样缓冲液混匀,取适量加样于凝胶点样孔内。
实验六 SDS聚丙烯酰胺凝胶电泳法—蛋白质的分子量测定【实验目的】1.掌握SDS—聚丙烯酰胺电泳法的原理。
2.学会用此种方法测定蛋白质的分子量。
【实验原理】SDS—聚丙烯酰胺凝胶电泳(SDS-PAGE)是对蛋白质进行量化,比较及特性鉴定的一种经济、快速、而且可重复的方法。
该法主要依据蛋白质的分子量对其进行分离。
SDS与蛋白质的疏水部分相结合,破坏其折叠结构,并使其稳定地存在于一个广泛均一的溶液中。
SDS—蛋白质复合物的长度与其分子量成正比。
由于在样品介质和聚丙烯酰胺凝胶中加入离子去污剂和强还原剂后,蛋白质亚基的电泳迁移率主要取决于亚基分子量的大小,而电荷因素可以被忽略。
SDS—PAGE因易于操作和广泛的用途,使它成为许多研究领域中一种重要的分析技术。
SDS是十二烷基硫酸钠(sodium dodecyl sulfate)的简称,它是一种阴离子表面活性剂,加入到电泳系统中能使蛋白质的氢键和疏水键打开,并结合到蛋白质分子上(在一定条件下,大多数蛋白质与SDS的结合比为1.4gSDS/1g蛋白质),使各种蛋白质—SDS复合物都带上相同密度的负电荷,其数量远远超过了蛋白质分子原有的电荷量,从而掩盖了不同种类蛋白质间原有的电荷差别。
这样就使电泳迁移率只取决于分子大小这一因素,于是根据标准蛋白质分子量的对数和迁移率所作的标准曲线,可求得未知物的分子量。
【实验材料】1.实验器材微型凝胶电泳装置;电源(电压200V,电流500mA);100℃沸水浴;Eppendorf管;微量注射器(50μl或100μl);干胶器、真空泵或水泵;带盖的玻璃或塑料小容器;摇床。
2.实验试剂⑴ 2mol/L Tris-HCl (pH8.8):取24.2g Tris, 加50ml蒸馏水,缓慢的加浓盐酸至pH8.8(约加4ml);让溶液冷却至室温,pH将会升高,加蒸馏水至100ml。
⑵ 1mol/L Tris-HCl (pH8.8):取12.1g Tris, 加50ml蒸馏水,缓慢的加浓盐酸至pH6.8(约加8ml);让溶液冷却至室温,pH将会升高,加蒸馏水至100ml。
变性梯度凝胶电泳变性梯度凝胶电泳是一种比较新型的电泳技术,它可以有效地用于分离和分析复杂的蛋白质组。
变性梯度凝胶电泳的原理是建立一种特殊的梯度,使用不同的离子强度来分离和收集分子。
由于离子强度是一个逐步变化的量,可以提供不同的环境条件,从而形成分子的各种簇,并将其运转至电泳柱的不同部位。
变性梯度凝胶电泳技术主要用于蛋白质分离和分析。
它可以更好地分离具有相似分子量的蛋白质,因为它可以根据溶解度,分子大小和芳香性等其他因素来调节电泳条件来实现有效的分离。
因此,变性梯度凝胶电泳技术可以用于解决较难分离的蛋白质,因为它可以根据空间分布来分辨蛋白质,并且实验结果更精确。
另外,变性梯度凝胶电泳可以应对大量的样品,更有效地分离出病毒感染的蛋白质,便于研究蛋白质的分子结构、活性和功能等相关信息。
变性梯度凝胶电泳的实验操作非常简单,准备一个用于变性梯度凝胶电泳的实验室,需要一台变性梯度凝胶电泳仪,并准备一种特殊的凝胶,可以在改变离子强度的基础上建立梯度。
然后将样品和标准样品混合后,放入变性梯度凝胶电泳仪中,开始电泳,随着离子强度的不断改变,蛋白质分子会呈现出不同的分布状态,最后在合适的时间内收集电泳带,完成变性梯度凝胶电泳的实验。
由于变性梯度凝胶电泳技术是一种比较新型的分析技术,所以也可以用于核酸分离和定量鉴定。
这种技术可以检测DNA和RNA的结构和数目,并可以调整梯度条件以更好地分离反应物,以达到最佳测定结果。
此外,变性梯度凝胶电泳技术还可以用于生物分子组学研究,以充分发挥它的作用。
总的来说,变性梯度凝胶电泳是一种新型的分析技术,可用于蛋白质和核酸的分离、定量和分析,以及组学研究,具有习惯性,灵敏度和效率高等特点,已经成为了当今生物分析中常用的分析手段。