基础有机化学 邢其毅 复习经验分享
- 格式:docx
- 大小:19.22 KB
- 文档页数:3

基础有机化学第三版邢其毅十、反应和反应机理有机反应:在一定的条件下,有机化合物分子中的成键电子发生重新分布,原有的键断裂,新的键形成,从而使原分子中原子间的组合发生了变化,新的分子产生。
这种变化过程称为有机反应(organic reaction)。
一级反应:在动力学上,将反应速率只取决于一种化合物浓度的反应称为一级反应。
二级反应:在动力学上,将反应速率取决于两种化合物浓度的反应称为二级反应。
按化学键的断裂和生成分类协同反应:在反应过程中,旧键的断裂和新键的形成都相互协调地在同一步骤中完成的反应称为协同反应。
协同反应往往有一个环状过渡态。
它是一种基元反应。
自由基型反应:由于分子经过均裂产生自由基而引发的反应称为自由基型反应。
自由基型反应分链引发、链转移和链终止三个阶段:链引发阶段是产生自由基的阶段。
由于键的均裂需要能量,所以链引发阶段需要加热或光照。
链转移阶段是由一个自由基转变成另一个自由基的阶段,犹如接力赛一样,自由基不断地传递下去,像一环接一环的链,所以称之为链反应。
链终止阶段是消失自由基的阶段,自由基两两结合成键,所有的自由基都消失了,自由基反应也就终止了。
离子型反应:由于分子经过异裂生成离子而引发的反应称为离子型反应。
离子型反应有亲核反应和亲电反应,由亲核试剂进攻而发生的反应称为亲核反应,亲核试剂是对正原子核有显著亲和力而起反应的试剂。
由亲电试剂进攻而发生的反应称为亲电反应。
亲电试剂是对电子有显著亲合力而起反应的试剂。
按反应物和产物的结构关系分类加成反应:两个或多个分子相互作用,生成一个加成产物的反应称为加成反应。
取代反应:有机化合物分子中的某个原子或基团被其它原子或基团所置换的反应称为取代反应。
重排反应:当化学键的断裂和形成发生在同一分子中时,会引起组成分子的原子的配置方式发生改变,从而形成组成相同,结构不同的新分子,这种反应称为重排反应。
消除反应:在一个有机分子中消去两个原子或基团的反应称为消除反应。

有机化学总结--邢齐毅work Information Technology Company.2020YEAR基础有机化学第一章、绪论有机化学—organic chemistry ;碳化合物— carbon compound KekuleA 提出碳四价理论。
Synthesis第二章、有机化合物的分类表示方法命名烃(hydrocarbon)R---rectus ; S ---- sinister第三章、立体化学第四章、烷烃自由基取代反应小环烷烃的开环反应:与氢反应属于自由基反应,与卤素和氢碘酸反应属于离子反应。
第五章、紫外光谱红外光谱核磁共振和质谱紫外和可见光谱(ultraviolet and visible spectrum) UV凡是能在一段光波内产生吸收的基团,就称为这一段波长的生色基团。
关键词:助色效应,助色基团,红移,蓝移(紫移),增色效应,减色效应。
Woodward Fieser规则。
红外光谱(Infrared Spectroscopy) IR实际吸收峰的数目少于振动自由度的数目:1.简并;2.偶极变化为零;3.仪器分辨率不高。
孤立甲基只在1380附近出现单峰,异丙基的双峰强度相等,三级丁基的双峰强度不等。
影响化学键和基团特征频率的因素:共振减三十,氢键减六十。
核磁共振(Nuclear Magnetic Resonance) NMR弛豫:纵向弛豫,横向弛豫。
偶合常数与化学位移不同,它不随外磁场的改变而改变。
磁等价化学一定等价,反之则不成立。
质谱(Mass Spectrum) MS分子被电子束轰击失去一个电子形成的离子称为分子离子。
分子离子峰的质量数要符合氮规则。
麦氏重排:γ氢转移。
第六章、脂肪族饱和碳原子上的亲核取代反应及β-消除反应关键词:诱导效应、共轭效应、超共轭效应。
Walden转换:构型转换。
影响亲核取代反应的因素:烷基的结构、离去基团的离去能力、试剂的亲核性及溶剂在反应中的作用。


第18章含氮芳香化合物芳香亲核取代反应18.1 复习笔记一、芳香硝基化合物硝基与苯环直接相连的化合物称为芳香硝基化合物(aromatic nitro compound)。
1.芳香硝基化合物的结构(1)根据分子中所含硝基的数目,可以分为一元、二元、三元或多元芳香硝基化合物。
一元芳香硝基化合物的通式为Ar-N02,与亚硝酸酯(nitrite)Ar0-N=0互为同分异构体。
(2)硝基的结构是对称的。
在芳香族硝基化合物中,硝基氮、氧上的p轨道与苯环上的p轨道一起形成一个更大的共轭体系。
硝基苯的结构如图l8-1所示。
图18-1 硝基苯的结构图2.芳香硝基化合物的物理性质(1)一元芳香硝基化合物都是高沸点的液体,多数是有机化合物的良好溶剂。
(2)最简单的芳香硝基化合物是硝基苯(nitrobenzene),它是淡黄色的油状液体,沸点211℃,具有苦杏仁味,不溶于水,而溶于多种有机溶剂中。
有毒。
(3)常用硝基苯做Friedel—Crafts反应的溶剂。
(4)二元或多元芳香硝基化合物一般为无色或黄色的固体。
3.芳香硝基化合物的重要化学性质(1)还原反应①在催化氢化或较强的化学还原剂的作用下,硝基可以直接被还原为氨基。
②在适当条件下用温和还原剂还原,则生成各种中间的还原产物,如亚硝基苯(nitrosobenzene)和苯基羟胺(phenylhydroxylamine)。
苯甲基羟胺在弱酸性及中性溶液中可以制备得到,但在强酸性还原体系中,由于很活泼,很容易转变成苯胺。
③硝基苯的最终还原产物是苯胺(aniline),苯胺是有机合成的重要中间体。
在酸性或中性条件中,硝基苯主要发生单分子还原反应(unimolecular reduction)。
各产物之间的关系如图18-2所示:图18-2 硝基苯单分子还原情况很难用还原的方法制备亚硝基苯,但它可通过苯胺或苯基羟胺的适当氧化来制备。
苯酚等活泼芳香族化合物与亚硝酸作用时可在羟基等活性基团的对位直接引入亚硝基。


第5章紫外光谱红外光谱核磁共振和质谱5.1 复习笔记一、紫外光谱(UV)1.紫外光谱的产生紫外光的波长范围是100~400 nm,它分为两个区段。
波长在100~200 nm称为远紫外区,这种波长能够被空气中的氮、氧、二氧化碳和水所吸收,因此只能在真空中应用,这个区域称为真空紫外区。
目前真空紫外区在有机化学中用途不大。
波长在200~400 nm 称为近紫外区,一般的紫外光谱是指这个区域的吸收光谱。
波长在400~800 nm范围的称为可见光谱。
常用的分光光度计一般包括紫外及可见两部分,波长在200~800 nm(或200~1000 nm)。
分子内部的运动有转动、振动和电子运动,相应地,分子具有转动能级、振动能级和电子能级。
电子能级的跃迁所需能量最大,大致在1~20 eV(电子伏特)之间。
根据量子理论,相邻能级间的能量差ΔE、电磁辐射的频率ν、波长λ符合下面的关系式ΔE=hν=h×c/λ式中h是普朗克常量,为6.624×10-34J·s=4.136×10-15eV·s;c是光速,为2.998×1010cm·s-1。
2.电子的跃迁有机化合物分子中主要有三种电子:σ电子、π电子和未成键的孤对电子(也称n电子)。
基态时,σ电子、π电子分别处在σ成键轨道和π成键轨道上,n电子处于非键轨道上。
仅从能量的角度看,处于低能态的电子吸收合适的能量后,都可以跃迁到任一个较高能级的反键轨道上。
跃迁时吸收能量的大小顺序为n→π*<π→π*< n→σ* <π→σ*<σ→π*<σ→σ*对于一个非共轭体系来讲,所有这些可能的跃迁中,只有n→π*。
跃迁的能量足够小,相应的吸收光波长落在近紫外-可见光区。
其他的跃迁能量都太大,它们的吸收光波长均在200 nm以下,无法观察到紫外光谱。
3.紫外光谱图紫外光谱图是以波长(单位nm)为横坐标,以化合物对电磁辐射的吸收强度或透过率为纵坐标的吸收曲线图。

第23章萜类化合物、甾族化合物和生物碱23.1 复习笔记一、萜类化合物1.萜类化合物的生物合成萜类(terpene)化合物是广泛分布于植物、昆虫、微生物等动植物体内的一类有机化合物。
在生物体内,萜类化合物是由乙酰辅酶A(简写为CH3COSCoA)转化而来的。
乙酰辅酶A的结构简式如下所示:(1)转化过程:首先乙酰辅酶A和二氧化碳结合转化为丙二酰辅酶A,后者再和一分子的乙酰辅酶A形成乙酰乙酰辅酶A,这个中间体再和一分子乙酰辅酶A进行羟醛缩合反应,就得一个六碳中间体,然后还原水解,产生萜的生物合成前体,3-甲基-3,5-二羟基戊酸。
乙酰辅酶A 丙二酰辅酶A 乙酰乙酰辅酶A六碳中间体3-甲基-3,5-二羟基戊酸(2)3-甲基-3,5-二羟基戊酸是生物合成的一个有效前体。
(3)由3-甲基-3,5-二羟基戊酸变为异戊二烯体系还需要失去一个碳原子。
经过腺苷三磷酸酯(ATP)的作用,两个羟基分步骤地进行磷酸化,然后失去磷酸,同时失羧,得到焦磷酸异戊烯酯(isopentenyl pyrophosphate)。
(4)由焦磷酸异戊烯酯再进行结合就可生成各种萜类化合物。
例如拢牛儿醇(geraniol)的生成过程如下所示:2.萜类化合物的结构组成和分类(1)结构组成①这些分子可以看作是两个或两个以上的异戊二烯分子,以头尾相连的方式结合起来的。
例如在对烷分子中,是由一个异戊二烯分子中的C-1和另一个异戊二烯分子的C-4′结合起来的:②异戊二烯规则(isoprene rule)。
现在已知:绝大多数萜类分子中的碳原子数目是异戊二烯五个碳原子的倍数,仅发现个别的例外。
(2)萜类化合物可以根据组成分子中异戊二烯单位的数目来分类。
碳原子数异戊二烯单位单萜l0 2倍半萜15 3二萜20 4三萜30 6多萜3.萜类化合物的实例(1)单萜从植物的花、叶、果皮、种子及树皮等中提取得到的具有芳香气味的易挥发的液体称为精油。
单萜类化合物是精油的主要成分之一。

第16章周环反应16.1 复习笔记一、周环反应和分子轨道对称守恒原理1.周环反应(1)定义:在化学反应过程中,能形成环状过渡态(cyclic transition state)的协同反应(synergistic reaction)统称为周环反应。
(2)协同反应是一种基元反应(elementary reaction)。
其含义是:在反应过程中,若有两个或两个以上的化学键破裂和形成时,都必须相互协调地在同一步骤中完成。
(3)周环反应具有如下的特点:①反应过程中没有自由基或离子这一类活性中间体产生。
②反应速率极少受溶剂极性和酸、碱催化剂的影响,也不受自由基引发剂和抑制剂的影响。
③反应条件一般只需要加热或光照,而且在加热条件下得到的产物和在光照条件下得到的产物具有不同的立体选择性(stereoselectivity),是高度空间定向反应。
④遵循微观可逆性原理。
(4)周环反应主要包括电环化反应(electrocyclic reaction)、环加成反应(cycloaddition)和σ迁移反应(σmigrate reaction)。
2.分子轨道对称守恒原理电环化反应在加热和光照条件下具有不同的立体选向性。
分子轨道对称性是控制这类反应进程的关键因素。
分子轨道对称守恒原理认为:化学反应是分子轨道进行重新组合的过程,在一个协同反应中,分子轨道的对称性是守恒的,即由原料到产物,轨道的对称性始终不变。
因此分子轨道的对称性控制着整个反应的进程。
二、前线轨道理论1.前线轨道理论的概念和中心思想(1)基本概念①最高占有轨道(HOMO):已占有电子的能级最高的轨道。
②最低未占有轨道(LUMO):未占有电子的能级最低的轨道。
③单占轨道(single occupied molecular orbital):有的共轭体系中含有奇数个电子,它的已占有电子的能级最高的轨道中只有一个电子。
用SOMO表示。
单占轨道既是HOMO,又是LUMO。

第25章新型有机合成方法25.1 复习笔记一、Heck反应1.定义卤代芳烃或烯烃与乙烯基化合物在过渡金属催化下形成碳碳键的偶联反应称为Heck 反应。
通常需要碱参与和在钯催化下进行。
2.机理Heck反应的机理如下所示:(1)零价或二价钯的催化剂前体被活化,生成能直接催化反应配位数少的零价钯。
(2)卤代烃对新生成的零价钯进行氧化加成。
这是一个协同过程,也是整个反应的决速步骤。
碘代芳烃反应最快,产率也较高,而且反应条件温和,时间短。
碘代、溴代以及氯代芳烃或烯烃的活性随着碳卤键的键能增加而递减,一般不使用氟代芳烃或烯烃进行Heck反应。
(3)第三阶段为烯烃的迁移插入,它决定了整个反应的区域选择性和立体选择性。
一般来说,烯烃上取代基空间阻碍越大,迁移插入的速率越慢。
如(4)整个循环的最后一步就是钯氢消除反应,生成取代烯烃和钯氢络合物。
后者在碱如三乙胺或碳酸钾等作用下重新生成二配位的零价钯,再次参与催化循环。
在此步骤中最重要的是反应的立体化学。
例如:3.应用Heck反应是合成带各种取代基的不饱和化合物最为有效的偶联方法之一。
对许多官能团如醛基、酯基、硝基等均有良好的兼容性。
利用分子内的Heck反应还可构筑稠环体系。
缺点:反应条件比较苛刻,需要比较严格的无氧操作,很多情况下对水也十分敏感。
二、Bergman环化反应1.定义共轭的烯二炔通过分子内环化生成1,4-苯双自由基或其类似物的一类环化反应称为Bergman环化反应,Bergman环化反应具有以下基本反应形式:启动该反应的关键反应条件是加热或者光照。
2.反应机理Bergman环化反应机理可以如下表示:在这个反应中,1,4-环已二烯作为氢供体使得1,4-苯双自由基不断地获得氢从而生成苯,苯不再可逆回到反应底物。
由于整个反应最终生成稳定的芳环体系,因此反应是放热的。
如果炔烃的取代基为丙基,则还会观察到三键被还原的产物出现,这说明反应与三键上的取代基有一定的关系:3.应用在构筑芳环体系、诱导烯烃的聚合以及药物化学等领域都有一定的应用价值。

基础有机化学邢其毅复习经验分享525616659(2011—06—20 14:16)下面的是比较广泛的一个经验当时写个别人的参考一下吧暑假的时候,很多人都会留在学校为考研积极备战.其实这个时候更大的是心理战,很多人回家呆久了之后,一想到正在学校努力学习的竞争对手就会有自己输在起点的感觉。
暑期在校虽然不能保证复习多少内容,但起码能给自己一个心理安慰,一个警示:我要考研了。
有一部分人还没最终确定自己要考哪里,但是已经有几个备选的学校或研究所,其实这都不是特别重要,毕竟可以先看看英语、政治等公共课,但有一点越早确定目标对后面的复习计划越有利,并且目标确定了不要轻易更改(我们班一回族同学当时要考上海XX,复习了挺长时间了才发现那的餐厅没有清真窗口,又临时改考上海**了)。
这个时候可以适当腾出些坐在自习室的时间,上网查一查到底哪个更适合自己,不仅从学科兴趣、师资、地理、以后的出路等等,不过好像更多的时候更多的人对某个要报考的地方是一见钟情……每一年考研过后都会有很多考的好的师兄师姐出来讲成功的经验,我觉得成功的经验不能听太多,尤其是讲自己如何在几天内突破某一科,非常轻松之类的话,那都是扯淡,即使真有也是运气成分……当一个人成功了,再讲他的成功之路时,经常会居高临下的面对一群崇拜者描述的很easy,因为他现在的心情就是很easy的,很难找到当时处于困境的感觉。
都说考研难,首先表现心理上。
虽然暑期时候大家见面都在抱怨考试内容多,但这都不是真正的抱怨.见面抱怨考试难是学生的通病,就好像街坊邻居见了面总爱问:“吃了没?”。
考研最考验一个人心理承受能力是在最后的那几天,现在没还必要细想这个。
(通常越临近考试越烦躁,越紧张,去年我们那个时候很多人就不在自习室学习了,嫌气氛压抑,受不了……话说回来了,紧张还是因为心里没底,要是每一科都认真复习了,习题认真做了,真题认真研究了,就会很淡定地盼着考试来证明自己,抑或是结束现在的痛苦生活呵呵。
卤代烃卤代烃的分类(本来是不想总结这一部分的,但后面做题的时候发现,题干中会出现,可能题目会做,但是你不知道这东西是什么。
所以,还是看看,有点印象比较好)按烃基结构分类1、饱和卤代烃2、不饱和卤代烃(这两类一定要分清楚,考试的时候,选择题可能不给你结构式,直接问。
)乙烯形卤代烃(与双键直接相连)烯丙型卤代烃(与双键的α-C相连)5、芳香卤代烃(苯型和苯甲型的,看一眼就记住了)卤代烃的命名这个自己看书(P218)卤代烃的结构1、饱和卤代烃。
α-C为sp3杂化。
C-X键为极性共价键,由于卤素的电负性大于碳原子,故而电子偏向卤素原子,那么α-C则会带有一部分正电荷,所以容易被亲核试剂进攻。
2、不饱和卤代烃一、与α-C以外的饱和碳原子相连。
与饱和卤代烃相似。
二、与α-C相连。
其生成的碳正离子(sp2杂化碳)由于双键的共轭,双键上的电子可以离域至α-C上以稳定正电荷,所以此类卤代烃活性要比饱和卤代烃的活性大。
三、与不饱和碳相连。
卤原子上的一对电子对参与不饱和键的共轭,故而使得C-X键或有双键的性质,所以反应活性最差。
此四类卤代烃的反应活性比较:烯丙型卤代烃>饱和卤代烃≈与α-C以外的饱和碳原子相连>与α-C以外的饱和碳原子相连。
卤代烃的构象一般情况下对交叉的构象最为稳定。
会有例外,做题的时候会遇到,但不多。
往往是根据机理来解释。
卤代烃的物理性质1、低级卤代烃为气体,高级卤代烃为固体。
2、所有卤代烃均不溶于水。
3、卤代烃的可极化性顺序:RI>RBr>RCl>RF。
和第七主族的顺序一致。
可极化性比较大,可转化成反应所需要的形状,有利于反应的进行。
故而RI、RBr、RCl易于进行反应。
(要是想对可极化性有一个充分的了解,可以去查看无机化学。
北师大版的在上册部分。
)卤代烃的反应(各种效应类的问题,后面会单独列出来)碳正离子(重点中的重点,好多题目不提,但是都会直接或者间接的考这个知识点,贯穿于整本基础有机化学)定义:含有一个只带有六电子的带正电荷的碳氢集团称为碳正离子。
第24章有机合成基础24.1 复习笔记逆合成分析理论与生源合成学说为现代有机合成设计思想的基石。
一、有机合成的要求和驱动力1.有机合成的要求(1)合成的反应步骤越少越好,每步反应的产率越高越好,以及原料越便宜越易得越好;(2)有机合成的目的是尽可能地选择最便宜最易得的原料,通过各种有机反应将原料化合物经“拼接”和“剪裁”最终转化成复杂的目标分子结构;(3)有机反应的作用是使原来分子中的某一个或几个化学键断裂并形成一个或几个新的化学键,从而完成由原料分子到目标分子的转换。
2.有机合成的驱动力(1)将各种新的有机反应应用于有理论或实用意义分子的合成中;(2)利用天然的或未被充分利用的原料合成各种具有应用价值的物质;(3)合成一些满足特定需求的特殊有机分子。
二、有机合成设计的基本概念1.起始原料、目标分子和逆合成分析(1)通过逆合成分析得到的最简单的化合物,即整个合成利用的第一个化合物称为起始原料(starting material,简写为SM)。
起始原料通常是一些商业化的产品或在自然界中大量存在的化合物。
在合成过程中需要各种试剂(reagent),通过试剂和起始原料或中间体反应可以生成各种新的中间体或目标分子。
试剂也是合成子的合成等价物。
(2)计划合成的分子称为目标分子(target molecule),通常用TM表示。
(3)逆合成分析是一种逻辑推理的分析过程。
它将目标分子按一定的规律通过切断或转换推导出目标分子的合成子或与合成子相对应的试剂。
逆合成分析用双线箭头“ ”表示。
其一般式可表达为:2.合成子、反合成子、合成等价物和切断(1)在分子中的化学键进行切断时所产生的分子碎片称为合成子或叫合成元(synthon)。
(2)合成子可以是正、负离子,也可以是电中性的。
正离子称为受体合成子(acceptor synthon),简称a,负离子称为供体合成子(donor synthon),简称d,电中性的合成子简写为e,其中的中性原子(radical)简写为r。
第15章碳负离子缩合反应15.1 复习笔记一、氢碳酸的概念和α氢的酸性氢碳酸的酸性强弱可用碳上的氢以正离子解离下来的能力表示,用pK a值来表示,值越小,酸性越强。
烷烃的酸性很弱。
烯丙位和苯甲位碳上的氢的酸性比烷烃强。
末端炔烃的酸性更强一些,环戊二烯亚甲基上的氢相对更活泼一些。
1.α氢的酸性与官能团直接相连的碳称为α碳,α碳上的氢称为α氢。
α氢以正离子解离下来的能力即为α氢的活性(酸性)。
通过测定α氢的pK a值或其与重氢的交换速率可以确定α氢的酸性强弱。
(1)α氢的酸性强弱取决于与α碳相连的官能团及其它基团的吸电子能力。
总的吸电子能力越强,α氢的酸性就越强。
一些常见基团的吸电子能力强弱次序排列如下:(2)α氢的酸性还取决于氢解离后的碳负离子(carbanion)结构的稳定性。
碳负离子的离域范围越大越稳定。
(3)分子的几何形状会影响α氢的酸性。
(4)与α氢的解离和介质的介电常数及溶剂化有关。
2.羰基化合物α氢的活性分析羰基的吸电子能力很强,因此羰基化合物的α氢都很活泼。
例如在NaOD—D20中,2-甲基环己酮的α氢均可被氘取代。
(1)羰基使α碳原子上的氢具有活泼性,是因为:①羰基的吸电子诱导效应;②羰基α碳上的碳氢键与羰基有超共轭作用。
(2)羰基旁所连的基团的不同导致了它们的α氢的活性也有差异。
可以从这些化合物本身的结构以及它们形成烯醇式后的结构来认识:含羰基化合物的α氢的酸性从大到小顺序:酰氯>醛>酮>酯>酰胺①在酰氯中,氯的存在增强了羰基对α碳的吸电子能力,从而也增强了α氢的活性。
同时氯的吸电子效应也使形成的烯醇负离子因负电荷分散而趋于稳定。
②在酯和酰胺中,烷氧基氧的孤电子对和氨基氮的孤电子对均可与羰基共轭而使体系变得稳定。
③酰胺氮上的孤电子对碱性较强,使共轭体系更加稳定,要解离α氢,形成烯醇负离子需要的能量更多,故酸性比酯还弱。
④当醛基中的氢被烷基代替后,由于烷基的空阻比氢大,从某种程度上讲阻碍了碱和氢的反应;另外,由于烷基对羰基具有给电子的超共轭作用,因此醛的α氢比酮的α氢活泼。
第26章有机材料、合成高分子和超分子26.1 复习笔记一、有机共轭材料有机光电功能材料包括小分子和高(大)分子化合物。
大分子化合物是有机共轭分子通过一定形式形成的聚集体。
新型有机共轭分子的合成是该领域创新的基础。
1.导电高分子材料在一定的条件下,有机共轭小分子或高分子材料(通俗地称为塑料)完全可以具有金属的性能,从而变成导体。
对于共轭高分子材料而言,它最简单的结构就是聚乙炔。
反式聚乙炔poly(transacetylene)(PA)的结构顺式聚乙炔的结构30多年来,已经发展了许多此类共轭化合物。
具有代表性的有:聚对苯聚对苯乙炔聚噻吩聚吡咯polypyrrole(PPy)聚苯胺polyaniline(PANi)聚芴polyfluorene(PF)这类材料是一种简单分子形成的长链聚合物或寡聚物,它是由重复的单元链段组成的,而每个单元链段则是由碳碳单键和不饱和共价键(双键或叁键)交替组成的。
这些共轭高分子材料大多具有半导体的特性,它们的导电性是各向异性的。
材料的电学性质是由它的电子结构决定的。
这些轨道与相邻的碳氢原子轨道键合构成了平面型的结构框架。
其余的未成键的p z轨道与这一分子平面垂直,它们相互重叠,形成了类似于一维状态碱金属的长程的π电子共轭体系。
量子力学的计算结果表明这种一维体系是不稳定的,容易发生导体到半导体的相变,也称为Peierls相变。
Peierls相变导致能量最低空轨道(LUMO)和能量最高占据轨道(HOMO)之间产生比较大的能隙,从而使相变后的聚合物不再是良导体。
掺杂是指通过氧化或还原的过程使导电高分子材料存分子结构内发生氧化或还原反应。
其作用机理如下:(1)真空状态(vacuum state):共轭链(undisturbed conjugation)(2)中性孤子(neutral soliton):自由基(free radical)(3)正孤子(positive soliton):碳正离子(carbonium)(4)负孤子(negative soliton):碳负离子(carbanion)(5)正极化子(positive polaron):阳离子自由基(radicalcation)(6)负极化子(negative polaron):阴离子自由基(radicalanion)(7)正双极化子(positive bipolaron):二价碳正离子(carbodication)(8)负双极化子(negative bipolaron):二价碳负离子(carbodianion)在掺杂状态下,会产生以上这些载流子,而载流子在材料中的迁移引起电导。
基础有机化学邢其毅复习经验分享
525616659(2011-06-20 14:16)
下面的是比较广泛的一个经验当时写个别人的参考一下吧
暑假的时候,很多人都会留在学校为考研积极备战。
其实这个时候更大的是心理战,很多人回家呆久了之后,一想到正在学校努力学习的竞争对手就会有自己输在起点的感觉。
暑期在校虽然不能保证复习多少内容,但起码能给自己一个心理安慰,一个警示:我要考研了。
有一部分人还没最终确定自己要考哪里,但是已经有几个备选的学校或研究所,其实这都不是特别重要,毕竟可以先看看英语、政治等公共课,但有一点越早确定目标对后面的复习计划越有利,并且目标确定了不要轻易更改(我们班一回族同学当时要考上海XX,复习了挺长时间了才发现那的餐厅没有清真窗口,又临时改考上海**了)。
这个时候可以适当腾出些坐在自习室的时间,上网查一查到底哪个更适合自己,不仅从学科兴趣、师资、地理、以后的出路等等,不过好像更多的时候更多的人对某个要报考的地方是一见钟情…… 每一年考研过后都会有很多考的好的师兄师姐出来讲成功的经验,我觉得成功的
经验不能听太多,尤其是讲自己如何在几天内突破某一科,非常轻松之类的话,那都是扯淡,即使真有也是运气成分……当一个人成功了,再讲他的成功之路时,经常会居高临下的面对一群崇拜者描述的很easy,因为他现在的心情就是很easy的,很难找到当时处于
困境的感觉。
都说考研难,首先表现心理上。
虽然暑期时候大家见面都在抱怨考试内容多,但这都不是真正的抱怨。
见面抱怨考试难是学生的通病,就好像街坊邻居见了面总爱问:“吃了没?”。
考研最考验一个人心理承受能力是在最后的那几天,现在没还必要细想这个。
(通常越临近考试越烦躁,越紧张,去年我们那个时候很多人就不在自习室学习了,嫌气
氛压抑,受不了……话说回来了,紧张还是因为心里没底,要是每一科都认真复习了,习题认真做了,真题认真研究了,就会很淡定地盼着考试来证明自己,抑或是结束现在的痛苦生活呵呵。
) 考研复习时有可能会跟当学期的课冲突,很多人选择逃课,我觉得上不上
课问题不大,关键是要下定决心:我这节课铁定不上了,就安心复习;或者是我这节课去好好听听老师都讲些什么。
忌讳想逃课又怕点名或者上课时候只低头看自己的考研书,因为这样基本都是在浪费时间,正所谓一心不可二用。
考研重在坚持,三天打鱼两天晒网肯定不行。
既然总觉的内容多,有时候还老爱忘,那就要连续复习,勤翻,勤看,勤写。
复习过程的放松也很必要,我说的不是一上午放松几回,而是一周,一个月的问题,我那时候基本上是两个星期打一次球,一个多小时就够了,因为老觉得时间不够呵呵。
关于复习的方法问题,每个人的习惯不一样,所以没有固定的模式。
只要自己觉得有效,觉得舒服、不枯燥、不烦躁就行。
我当时喜欢拿整个的时间来复习某一科,比如一上午只看有机,一晚上只看物化。
其实我巴不得一整天都看有机,不过那样确实不太合理呵呵。
而有的人喜欢一天里把所有的科目都照顾一遍。
根据我个人的经验,有机需要连续看的时间长一点才
能看的透彻,一个小时肯定是不行的;物化也是这样甚至需要更久(如果你也考物化的话)。
复习方法的问题可以边复习边摸索。
有机化学的知识点比较多,但总起来说有机化学用来考试的题型不外乎以下几种: 1、命名。
因学校而异。
医工院考得不多可以放到过了十二月再开始看 2、选择题:这部分题主
要是基础知识,常见的有比较题(酸碱性、芳香性、稳定性、反应活性、氨基酸等电点等)。
3、完成反应:大多在习题集上能找到类似的反应。
要注意周环反应,还要辨清相
似的反应。
4、机理:反应机理有几个经典的模式,像SN1、SN2亲核取代,构型翻转与
保持,Claisen缩合与逆Claisen缩合,[3,3]- σ迁移,还有一些开环、关环机理的组合。
机理题中一般默认H+和H2O是随手捏来的。
做机理题忌眼高手低,需认真体会每一步骤的电子转移与结构变化,因为机理题是按步骤给分的。
5、合成:合成是有机化学取得高分
的关键。
一般都会有长链烷烃化合物、羰基化合物的合成,杂环化合物的合成也是一大热点。
做合成题的关键是基础反应了然于胸、运用得当,这几个字看起来容易做起来却比较耗时。
每一道合成题的路线必然都是最常见反应的组合,如果你的答案自己都觉得很勉强,那多半是有问题的。
6、结构推断:这部分10-30分不等。
因为研究生做实验,结果的判
定依靠的就是核磁、质谱等,因此结构推断就几乎等于波谱题(一般只涉及红外和核磁,其他的波普不需要看)。
这部分最难不过是同时给出氢谱、碳谱、质谱、红外、紫外,然后推断结构。
注意写出化合物质谱的裂解途径也是常见的题型。
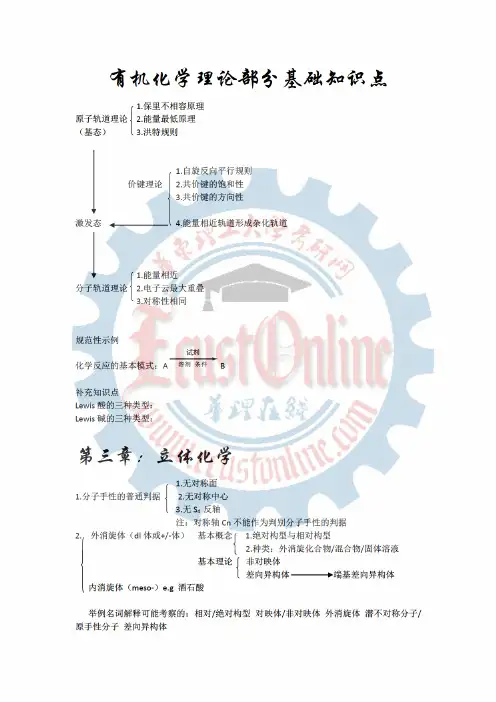
7.举例名词解释人名反应
掌握了考试的题型,复习起来就会有很大的针对性。
当然不排除一些个别的学校的个别新颖题型,虽说万变不离其宗,但多看一点怪题、偏题,确实能从中得到不少启发。
就《有机化学》邢其毅三版来说,比较重要的有: 1、烷烃的亲核取代,β-消除。
2、有机金属
化合物。
主要是格式试剂的应用范围。
基团选择性反应,一般只出选择题。
3、烯烃、炔
烃的亲电加成,包括马氏/反马氏等规则,以及加成机理。
其实硬记一个反应的产物是什么,不如去理解一类反应的机理,因为这样可以起到举一反三的效果。
4、苯环上的反应。
包括芳香亲电取代(硝化、卤化、磺化等),F-C反应等。
这里还包括芳香性的判断,综
合杂环部分来看,要分清在什么情况下不饱和键的π电子才算数(需环状连续共轭体系),一个不饱和键算几个π电子(双键算2个,三键仍然算2个,因为三键的两个p轨道是垂直的,有且只有一个p轨道与共轭不饱和键的p轨道平行),杂原子的孤电子对算不算作这里的π电子(吡咯的N原子算,吡啶的N原子则不算,算与不算要看哪种方式
能形成4n+2电子结构,因为4n+2的芳香结构是稳定的。
一个环状结构不符合4n+2电子
结构,它就是反芳香结构,是不稳定的),了解了这几点就比较容易判断了。
5、醇、醛、酮、羧酸四类化合物都比较重要。
把它们放在一起是因为它们之间的转化往往仅需一步,互为前体,并且这4类化合物的性质很活泼,代表反应很多,都是经常考到的。
每一章的最后都会讲该类化合物的制备,看这一部分对合成题是个不错的补充。
6、缩合反应。
非
常重要的一章。
上述6种题型都出现过缩合反应,尤其是机理与合成。
7、周环反应。
这
部分只需看一遍内容,很少会考。
但最好好好看,因为分子轨道理论的东西理解的越深刻,
对前边的知识点理解的也越深刻。
8、含氮化合物。
这部分涉及到胺和含氮芳香化合物,
经典反应比较多,会经常考到。
所谓经典反应、重点反应就是经常考的,试卷上出现很频繁的反应,习题做多了自然就分的清那是重点。
9、杂环。
杂环内容比较多,但常考的无
非是反应时,杂环上的区域选择性,再有就是杂环的合成,反应不多,但是较难理解,多看就边就好了。
10、其余的一些糖、氨基酸、甾体之类的有时间就看,实在没时间就算。
11、波谱题。
红外、核磁爱出题,规律性还比较明显。
核磁基本就是考解谱。
差不多整个有机化学就这些内容。
一般只需要看前20章,21 22看一下基本的概念就可以了。