电化学 第6章 气体电极过程
- 格式:doc
- 大小:1.49 MB
- 文档页数:15






思考题3. 从理论上推导电化学方程式(巴特勒-伏尔摩方程),并说明该理论公式与经验公式的一致性。
答:电化学极化处于稳定状态时,外电流密度必定等于(j j =v w),也就是等于电子转移步骤的净反应速度(即净电流密度j 净)。
由于电子转移步骤是控制步骤,因而j 净也应是整个电极反应的净反应速度。
这样,根据电子转移步骤基本公式,易得稳态电化学极化时电极反应的速度与电极电位之间关系。
即j=j 净。
将公式 j 净= 0F F RT RT j e e a b j j -D D 轾犏-犏臌 代入上式,则 0F F RT RT j j e e a b j j -D D 轾犏=-犏臌(1) 式(1)就是单电子电极反应的稳态电化学方程式,也称巴特勒-伏尔摩方程。
若电极反应净速度预用正值表示时,可用j c 代表阴极反应速度,用j a 表示阳极反应速度,将式(1)分别改写为0c c F F RT RT c j j j j e e a b h h -轾犏=-=-犏臌v w (2) 0a a F F RT RT a j j j j e e b a h h -轾犏=-=-犏臌w v (3)当过电位很大时,相当于双曲线函数x 值很大,即式(2)中有如下关系c c F F RT RT e ea b h h -?可以忽略(2)中右边第二个指数项,即 0c F RT c j j e a h -» (4)两边取对数02.3 2.3log c c RT RT j j F Fh a a =-+ (5) 同理,对于阳极极化为0a F RT a j j eb h -» (6) 02.3 2.3log a a RT RT j j F Fh b b =-+ (7) 式(5)和式(7)即为高过电位时巴特勒-伏尔摩方程近似公式。
与电化学极化的经验公式——塔菲尔公式(log a b j h =+)相比,可看出两者是完全一致的。
这表明电子转移步骤的基本动力学公式和巴特勒-伏尔摩方程的正确性得到了实践的验证。




电化学原理试题(1)第六章电化学极化1. 简述三种极化的概念,哪⼀种极化严格来讲不能称为极化。
电化学极化:当电极过程为电化学步骤控制时,由于电极反应本⾝的“迟缓性”⽽引起的极化。
浓度极化:当电极过程由液相传质步骤控制时,电极所产⽣的极化。
电阻极化:由电极的欧姆电阻引起的电位差。
电阻极化严格来讲不能称为极化2. 简述电化学极化最基本的三个动⼒学参数的物理意义。
1) 对称系数:电位偏离形式电位时,还原反应过渡态活化能改变值占F 的分数。
物理意义:反应改变电极电位对还原反应活化能的影响程度;(1—)反应改变电极电位对氧化反应活化能的影响程度。
对称系数是能垒的对称性的变量,是由两条吉布斯⾃由能曲线的斜率决定的,⽽且曲线的形状和斜率是取决于物质的化学键特性。
在CTP动⼒学中,可以⽤来推测过渡态的构型,研究电极反应的放电机理。
2)电极反应标准速率常数K:当电极电位等于形式电位时,正逆反应速率常数相等,称为标准速率常数。
物理意义:在形式电位下,反应物与产物浓度都为1时,K在数值上等于电极反应的绝对反应速度。
a.度量氧化还原电对的动⼒学难易程度;b体现电极反应的反应能⼒与反应活性;c.反应电极反应的可逆性。
3)交换电流密度J。
:在平衡电位下,氧化反应和还原反应的绝对电流密度相等,称为交换电流密度。
物理意义:a.度量氧化还原电对的动⼒学难易程度;b体现电极反应的反应能⼒与反应活性;c.反应电极反应的可逆性;d.表⽰平衡电位下正逆反应的交换速度。
3.为什么电极电位的改变会影响电极反应的速度和⽅向?4.写出Butler-Volmer公式在不同过电位范围下的近似公式。
5.简述J0对电极性质的影响。
6. J0描述平衡状态下的特征,为何它却能说明电化学动⼒学中的⼀些问题?7. 如何⽤稳态法测量三个动⼒学参数。
8. 在谈到⼀个CTP的不可逆性时,我们有时说它是过电位较⼤,⽽有时⼜说它是电流密度较⼩,这两种说法有何区别和联系?9.电解H2SO4⽔溶液,Ni阴极的过电位为0.35 V。
第六章 化学反应与能量李仕才第三节电解池 金属的电化学腐蚀与防护考点二 电解原理的应用 多池组合装置1.电解饱和食盐水 (1)电极反应阳极:2Cl --2e -===Cl 2↑(反应类型:氧化反应), 阴极:2H ++2e -===H 2↑(反应类型:还原反应)。
(2)总反应方程式:2NaCl +2H 2O=====电解2NaOH +H 2↑+Cl 2↑。
离子方程式:2Cl -+2H 2O=====电解2OH -+H 2↑+Cl 2↑。
(3)应用:氯碱工业制烧碱、氢气和氯气阳极:钛网(涂有钛、钌等氧化物涂层)。
阴极:碳钢网。
阳离子交换膜:①只允许阳离子通过,能阻止阴离子和气体通过。
②将电解槽隔成阳极室和阴极室。
2.电解精炼铜3.电镀铜4.电冶金利用电解熔融盐的方法来冶炼活泼金属Na、Ca、Mg、Al等。
判断正误(正确的打“√”,错误的打“×”)1.用铜作阳极、石墨作阴极电解CuCl 2溶液时,阳极电极反应式为2Cl --2e -===Cl 2↑。
( × )2.电解MgCl 2溶液所发生反应的离子方程式为:2Cl -+2H 2O=====电解Cl 2↑+H 2↑+2OH -。
( × )3.氯碱工业用阳离子交换膜把阴极室和阳极室分开。
( √ )4.Cu +H 2SO 4===CuSO 4+H 2↑可以设计成电解池,但不能设计成原电池。
( √ ) 5.粗铜电解精炼时,若电路中通过2 mol e -,阳极减少64 g 。
( × ) 6.电解冶炼镁、铝可电解熔融的MgO 和AlCl 3。
( × )1.粗铜中含有的相对活泼的物质也会失去电子,不活泼的金、铂形成阳极泥,而溶液中只有Cu 2+得到电子生成Cu ,故c(Cu 2+)将减小,并且阴极增重质量,不等于阳极减小的质量。
2.电镀时,阳极(镀层金属)失去电子的数目跟阴极镀层金属离子得到电子的数目相等,因此电镀液的浓度保持不变。
徐州工程学院教案徐州工程学院教案纸峰电流ip 可表示为:(1)式中K、n、D、v、A 和 c 分别为常数、电子转移数、扩散系数、电压扫描速率、电极面积和被测物质浓度。
由式(1) 可见,峰电流与被测物质浓度、扫描速率等因素有关。
从循环伏安图可确定氧化峰峰电流i pa和还原峰峰电流i pc,氧化峰峰电位φpa和还原峰峰电位φpc值。
对于可逆体系,氧化峰与还原峰的峰电流之比为:(2)氧化峰与还原峰的峰电位之差为:(mV) (3)条件电位:由此可判断电极过程的可逆性。
铁氰化钾离子-亚铁氰化钾离子氧化还原电对的标准电极电位:三、仪器及试剂仪器:LK2005A电化学工作站(CHI);铂片电极2 支;饱和甘汞电极1 支。
试剂:2.0×10-2mol·dm-3铁氰化钾标准溶液,1.0mol·dm-3的氯化钾溶液,蒸馏水。
铬酸洗液:20g的K2Cr2O7,溶于40mL水中,将浓H2SO4360mL徐徐加入K2Cr2O7溶液中(千万不能将水或溶液加入H2SO4中),边倒边用玻璃棒搅拌,并注意不要溅出,混合均匀,冷却后,装入洗液瓶备用新配制的洗液为红褐色,氧化能力很强,当洗液用久后变为黑绿色(可加入固体高锰酸钾使其再生),即说明洗液无氧化洗涤力。
四、操作步骤电极可逆性判断在电解池(小烧) 中放入少(20mL ) 2.0×10-2mol·dm-3铁氰化钾标准溶液,1.0mol·dm-3的氯化钾溶液,插入2 支铂电极和 1 支饱和甘汞电极(取下橡胶帽)。
其中一支铂电极作指示电极,另一支铂电极作辅助电极,饱和甘汞电极作参比电极。
以扫描速率,从+0.30 ~ -0.5 V 扫描,记录循环伏安图并判断电极可逆性。
存盘并记录ipa、ipc 和φpa、φpc 的值。
【铂片电极的处理:如上述判断出电极不可逆,则用铬酸洗液浸泡10 ~ 20 min 进行处理,然后用蒸馏水清洗,备用。
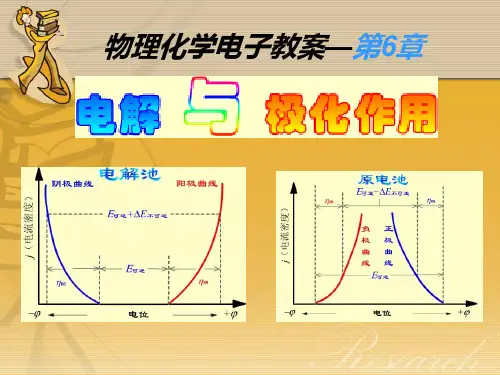
5.电极过程和电极过程动力学5.1电化学装置的可逆性:化学反应可逆性;热力学上可逆性5.2电极的极化5.3电极过程的控制步骤:电极反应的特点;电极反应的控制步骤5.4电荷转移动力学方程5.5交换电流密度与电极反应速度常数5.6稳态极化时的电极动力学方程5.7浓差极化及其电机动力学方程5.8化学极化分解电压E分:在可逆情况下使电解质有效组元分解的最低电压,称为理论分解电压(V e)。
理论分解电压是阳极平衡电极电位(εe(A))与阴极平衡电极电位(εe(K))之差。
Ve=εe(A)- εe(K)(10 - 5)当电流通过电解槽,电极反应以明显的速度进行时,电极反应将会明显偏离平衡状态,而成为一种不可逆状态,这时的电极电位就是不平衡电位,阳极电位偏正,阴极电位偏负。
这时,能使电解质熔体连续不断地发生电解反应所必需的最小电压叫作电解质的实际分解电压。
显然,实际分解电压比理论分解电压大,有时甚至大很多。
实际分解电压简称分解电压(V),是阳极实际析出电位(ε(A))和阴极析出电位(ε(K))之差。
V=ε(A)- ε(K)(10 - 6)当得知阴、阳极在实际电解时的偏离值(称为超电位)就可以算出某一电解质的实际分解电压。
分解电压符合能斯特方程,可以表示为如下形式:式中E i,E0分别表示实际和标准状态下组元i的分解电压;a i__组元的活度;n i __组元在熔盐中的化合价;F __ 法拉弟常数;可以看出,温度和电解质组成均会影响分解电压电极极化电解时的实际分解电压比理论分解电压要大很多,这是由于电流通过电解槽时,电极反应偏离了平衡状态。
通常将这种偏离平衡电极电位的现象称为极化现象。
电解过程实际分解电压和理论分解电压之差称为超电压。
⏹电解电极反应一般包含1:☐(1)反应离子由熔体向双电层移动并继续经双电层向电极表面靠近。
这一阶段在很大程度上靠扩散实现,扩散则是由于导电离子在熔体和双电层外界的浓度差别引起的。
☐(2)反应离子在电极表面进行电极反应前的转化过程,如表面吸附等;☐(3)在电极上的电子传递- - 电化学氧化或电化学还原反应;☐(4)反应产物在电极表面进行反应后的转化过程,例如自电极表面的脱附,反应产物的复合、分解和其它化学反应;☐(5)反应产物形成新相,或反应产物自电极表面向电解质熔体的传递。
第6章 气体电极过程所谓气体电极过程是指涉及气体的电极反应。
换句话说,反应物或产物为气体的电极反应就是气体电极过程。
如工业生产中电解制备H 2和Cl 2。
222Cl e Cl →---再如:--↔++OH O H e O 42422也是气体电极过程。
研究气体电极过程的主要目的是了解气体电极过程的规律,控制反应的进行,使其为科研和生产服务。
比如在电解法制备氢气中,我们可以设法降低氢析出过电位(如选择电极材料等),从而节约能源。
再如在电池工业中,充电过程中正极要有2O 析出,该反应都是副反应,对电池充电有害,我们通过研究可以设法提高2O 的析出过电位,从而提高电池的充电效率。
同样在电镀技术中,负极常常伴随着2H 的析出,由于存在该反应,使镀液体系的电流效率下降。
若能设法提高2H 的析出过电位,则电流效率则可以提高,从而节约能源。
还有就是22O H -燃料电池、Air Al ,Air Zn --电池等等都涉及气体电极过程,这方面例子很多,这里就不再一一列举了。
在气体电极过程中,研究得比较多,比较透彻的是氢、氧电极过程。
尤其是氢电极过程研究得最多,重现性好,人们认识也比较一致,有关氢电极过程的一些理论也是比较成熟的。
而氧电极研究的也不少,但认识不一致,提出几十种机理。
因此本章以氢电极为重点,氧电极也做一定介绍。
由于气体电极过程一般都涉及表面转化问题(或易于生成新相),大都涉及在电极上的吸附、吸附态中间粒子。
故先讨论吸附问题:6.1 氢原子和氧在电极上的吸附本节主要介绍吸附的方式、研究吸附的方法和吸附量的求出。
氢通常是以原子形式吸附的,而氧的吸附则是很复杂的,故不特指氧原子。
(可能形成222222O H HO HO O OH OH O O 、、、、、、、----…,ϕ不同,也可几种形式同时存在)。
氢原子吸附主要在Ni Fe Pd Pt 、、、等过渡金属表面上,而在Zn Cd Pb Hg 、、、等金属表面上从未发现过较大量的吸附氢原子。
而在所有金属表面上均可以出现氧的吸附和氧化物层。
目前对电极表面上含氧粒子的性质还没有一个满意的检定方法,因此所谓“氧的吸附”其含义往往并不完全清楚。
--+↑→+OH H e O H 222226.1.1气体吸附种类气体吸附有两种:① 物理吸附....:气体分子的吸附,是由分子间的范德华力引起的,只能在低温下实现。
② 化学吸附....:吸附物质与电极之间的相互作用与生成化学键相似。
这种属于化学吸附的氢或氧的吸附态,既可以通过气体吸附(化学作用)形成,也可通过电化学反应生成: MH e H M ↔++-+ (Had MH = 吸附氢原子) 即+H 的还原,生成 Had-+++-↔+e H O M O H M 222 (O M -吸附氧)--++-→+e O H O M M OH 222即O H 2的氧化生成O M - (碱性溶液中-OH 氧化)这种吸附态的形成伴随着电子得失。
在气体电极过程的历程中,一般都有此类电化学吸、脱附步骤,它对整个反应的动力学规律往往有影响,需要加以研究。
6.1.2 气体吸附的研究方法循环伏安法(Cyclic Voltammetry )该方法是研究电极行为的常用方法之一,该方法具有简单,易行,快捷等优点。
(1)线性扫描电位法首先介绍一下电位扫描法。
所谓线性电位扫描法(Linear Sweep Potential)是控制ϕ在一定范围内以一定的速度做线性变化,即使const dtd =φ,这时电极发生极化,极化电流将随ϕ变化,记录下i 的变化,就得到了极化曲线。
若扫描速度足够小,就是恒电位法。
相当于给定一个1ϕ,测1i ; 给2ϕ,测2i ,…画出曲线。
注意:这里虽然ϕ随t 变化,但却相当于稳态法,而不是暂态法。
(速度慢→稳态) 暂态法:恒i (或恒ϕ),用响应速度快的仪器测出非稳态扩散时t -ϕ(或t i -)曲线的方法。
这种测量的特点是可消除浓差极化影响,测出电化学步骤的动力学参数。
稳态法:仪器响应慢,恒i (或恒ϕ)得到稳态下的极化ϕ(或i )。
自变量变化,但当你测出对应的因变量时已是稳态值。
线性ϕ扫描:记录仪响应慢,i 的变化。
曲线上任一点i 都是某一ϕ下的稳态i ,所以该法又有人称为“动”电位扫描或“恒ϕ扫描”。
该方法可观察较宽ϕ范围内电极过程的变化,信息量较大。
(2)循环伏安法循环伏安法(V-A ):是线性ϕ扫描法的一种。
它是使ϕ变到一定值后再改变方向做反向变化,循环一周(或几周)。
t -ϕ曲线为三角波,ϕ变化一个周期:前半周若对应阳极氧化(从e ϕ开始),后半周显然将对应阴极还原(前述过程的逆过程)。
该方法便于了解电极反应的全貌,并能发现一些半周期扫描看不到的现象。
i )测量关于测量原理简单说:将三角波信号输入恒电位仪,恒电位仪控制WE (Vs.RE )的电位按输入信号变化,WE 发生变化。
极化回路中有i 通过,且i 随ϕ变。
用X-Y 记录仪将极化曲线记录下来(ϕ输入X 轴,i 输入Y 轴),即完成测量。
Pt 电极在42SO H 溶液(2.3M )中的循环伏安曲线如图7-6,RE 为同一溶液中的氢电极。
1)横轴上半枝阳极曲线。
随着ϕ→(+),氢脱附、双层充电、氧吸附。
2)横轴下半枝阴极曲线。
随着ϕ→(-),氧脱附、双层充电、氢吸附。
也可以分为三个区:氢区,双层区,氧区。
ii )曲线分析从上述曲线可看到的现象:① 氢的吸、脱附峰对应的电位基本相同,说明吸、脱附基本可逆。
MHe M H ⇔++-+Pt 是低2H η金属,催化性好,0i 大,可逆性好,在e ϕ附近反应基本可逆。
图6-4 循环伏安法电位-时间曲线图6-6 气体吸脱附的循环伏安曲线[42SO H 溶液(2.3M )]② 氢的吸脱附均为两个峰,说明氢在Pt 上吸附的键能不同。
较负的ϕ峰(第I 峰)下是弱吸附的H 的吸脱附,而较正的ϕ峰(第II 峰)下是强吸附H 的吸脱附。
就是说,吸附或脱附大致分为两步进行:刚开始吸附时(即H θ较小时),H 原子与金属结合较强(牢固),这部分Had (吸附极化小,脱附极化大)不易脱附,在+0.2~0.4V 时脱附。
随着H θ↑,利于吸附的点已被占据,且相互排斥力↑,吸附逐渐变得困难了。
进一步吸附的H 与金属结合较弱,易脱附,在0.2V 之前完成(极化小)。
这种强弱之分是有实验根据的:θ<0.5时,吸附热为10~15千卡/mol (放热多,键强);θ≥0.5时,吸附热为7~8千卡/mol 。
进一步:为何先吸附结合强,后吸附者结合弱?可能是电极表面不均匀造成的。
先吸附的占据了能量较低的位置,稳定。
后吸附者只能占据能量较高的位置(不太稳定)。
通常以ϕ峰为反应的特征值,有一ϕ峰对应一反应。
对阳极反应,ϕ峰越负,极化越小,可逆性好。
对阴极反应,ϕ峰向正偏移,极化小,可逆性好。
[阴极还原,ϕ→(-)变,先吸附强者,ϕ较正,C η小,后吸附者弱,ϕ较负。
与阳极反应对应ϕ峰相同,易吸附者,在C η较小时即吸附,而不易吸附者在C η较大时才吸附,但已不能占据有利位置,故吸附弱]。
另外:吸、脱附曲线下面积(对应It Q =)应相等。
③氧区:吸、脱附电位不等,脱吸ϕϕ>,该过程不可逆(相差越多,不可逆性越大)。
另:阳极极化时的电量比阴极极化时大,C A Q Q >。
即吸附氧消耗电量大,而氧脱附时,消耗电量小,说明吸附的氧未完全脱附,有残留的O M -,也说明了过程的不可逆性。
6.2 氢析出反应的基本规律及可能机理在气体电极过程中,对于氢析出反应研究得最透彻,数据重现性较好,关于反应机理认识也较一致,因此首先将氢析出过程的研究成果加以介绍。
首先,人们经大量试验总结出了关于该反应的基本规律,从这些基本规律出发提出了若干可能的反应历程,然后再经大量试验,充分验证其中哪一个是正确的。
因此我们还是从介绍基本试验规律入手,逼近反应实质。
6.2.1 反应规律1、大多数金属上(反应载体,惰性电极)的2H 析出反应均须在高C η下进行,即符合Tafel 方程FRT b i b a ib ac C αη303.2lg lg 0=-=+=0lgii b C =η 010i i ≥时, V n C /118.0≥η 这一事实早在1905年即由Tafel 发现了。
2、不同体系(不同电极or 电液),a 、b 值不同。
举例:体系a b 421/SO H N Pb 1.56 0.110 421/SO H N Hg1.415 0.113 HCl N Hg 1/ 1.406 0.116 423.1/SO H N Cd1.40 0.12 421/SO H N Zn 1.24 0.118 HCl N Sn 1/ 1.24 0.116 HCl N Ag 1/ 0.95 0.116 HCl N Fe 1/ 0.70 0.125 HCl N Cu 2/ 0.80 0.125 HCl N Pt 1/ 0.10 0.13 422/SO H N Pd0.260.123、b 值变化范围较小,0.11~0.13(0.12附近) cc F RT b αα059.0303.2==T 一定时,表现了C η时的活化能影响程度,b 值相近。
说明ϕ对2H 析出过程活化能影响大致相同,若在所涉及的电势范围内表面状态发生了变化如表面氢化,则b 值较高>140mV 。
4、不同电极体系,a 值相差较大 0.1~1.5① a 的物理意义:21-⋅=cm A i 时过电位的数值,即i =1时,a C =η(当然0i 单位也是2-⋅cm A )a 大,C η往往也大。
② 0lg i b a -=,已知0i 越大,反应可逆性越好,故也可用a 值判断或比较可逆性好坏:0i 越大,a 越小,可逆性越好,也表现了对反应的催化能力很大,则在该电极上2H 析出只需较小的过电位;反之0i 越小,a 越大,可逆性越差,说明该电极反应的催化能力小,则该电极上2H 析出常需较大过电位。
③ 因此,可按a 值大小,将常用的电极材料分为三类:高2H η金属:(a=1.0~1.5)Tl Sn Bi Sb Zn Hg Cd Pb 、、、、、、、.( ta ) 中2H η金属:(a=0.5~0.9)Au Ag Cu Ni Co Fe 、、、、、.低2H η金属:(a=0.1~0.3)Pd Pt 、.所谓氢过电位(2H η)即指2H 析出的还原反应所需阴极过电位。
6.2.2 氢析出反应的可能机理氢析出过程(2H e H →+-+的阴极过程)基本的动力学特征是η与i 之间成半对数关系,(大多数金属上是如此),即有i b a lg +=η (Tafel 关系),大家知道电化学步骤为控制步骤时,也有此Tafel 关系,随后转化步骤为控制步骤时,也有此Tafel 关系。