第二章 价键理论、晶体场理论
- 格式:doc
- 大小:254.00 KB
- 文档页数:40


第四章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论(Valence bonding theory VBT)②晶体场理论(Crystal field theory CFT)③分子轨道理论(Molecular orbital theory MOT)第一节价键理论由L. C. Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化(Hybrid orbital)及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
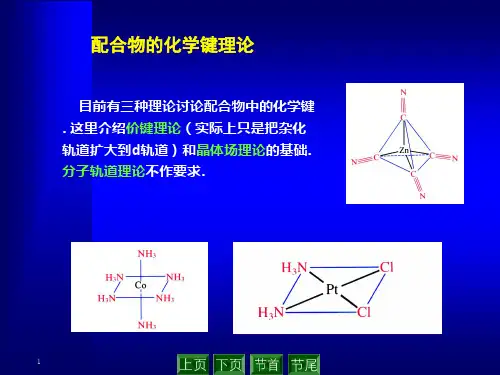
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp 3、sd 3杂化 四面体顶点 Ni(CO)4COCOOCCOsp 2、sd 2、dp 2、d 3杂化 三角形顶点 [AgCl 3]2-Cl ClClAgdsp 2、d 2p 2 杂化 正方形顶点 [PtCl 4]2-ClClClClPtd 2sp 3杂化 八面体顶点 [Fe(CN)6]4-CNNCFeNCCNsp杂化直线型[AgCl2]-二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
O.+xf = x(p x) = ?类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
2、如何判定原子轨道波函数的对称类型例:[HgI 3]− (D 3h 群)平面三角形III HgD 3hE 2C 33σv 11111-12-10z(x, y)x 2+(x 2(x3C 2σh 2S 3A 1'A 2'E'A 1"A 2"11111-12-101111-1-111-1-1-112-1-21E"A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A2″:p zE″:(d xz、d yz)3、轨道杂化方案步骤:A、以一组杂化轨道集合作为分子所属点群表示的基,写出群的表示。




配位化学讲义第四章(1)价键理论、晶体场理论第三章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valencebond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[Fe(CN)6]4-sp杂化直线型[AgCl2]-二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A 2″:p zE″:(d xz、d yz)2、σ轨道杂化方案1)四面体分子AB4(Td)[CoCl4]2-以四个杂化轨道的集合作为分子点群(Td)表示的基,确定该表示的特征标:r1r4r2r3恒等操作,χ(E)=4 C3操作,χ(C3)=1对C2、S4和σd用同样方法处理,得T d E 8C3 3C2 6S46σdΓ 4 1 00 2约化:T d E 8C3 3C2 6S4 6σdA1 1 1 1 11A2 1 1 1 -1 - 1E 2 -1 2 00 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -11 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+6×(-1)×2]=0a(E)=1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+6×0×2]=0a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24 [3×4+8×0×1+3×(-1)×0+6×(-1)×0+6×1×2]=1约化结果Γ=A1+T2由特征标表:A1T2s(p x、p y、p z)(d xy、d xz、d yz)可有两种组合:sp3(s、p x、p y、p z)、sd3(s、d xy、d xz、d yz)* 以一组杂化轨道为基的表示的特征标的简化计算规则:①不变(1)②改变符号(-1)③与其他函数变换(0)2)再以[CdCI5]3-三角双锥(D3h)为例:41325D3h E 2C33C2σh2S3 3σvΓ 5 2 13 0 3约化结果:Γ= 2A1′+A2〞+E′A1′A2〞E′s p z (p x、p y)d z2(d xy、d x2-y2)两种可能的组合:(s、d z2、p z 、p x、p y)( s、d z2、p z、d xy、d x2-y2)3)[HgI3]- ( D3h)123D3h E 2C3 3C2σh2S33σvΓ 3 0 13 0 1约化得:Γ=A1′+E′A1′E′s (p x、p y)d z2(d xy、d x2-y2)可能的组合有:(s、p x、p y)、(s、d xy、d x2-y2)、(d z2、p x、p y)、(d z2、d xy、d x2-y2)4)平面AB4型分子(D4h)例:[PtCl4]2-C2′C2″D4h E 2C4(C41,C43) C2(C42) 2C2′2C2″i 2S4σh 2σv2σdΓ 4 0 0 20 0 0 4 2 0约化得:Γ=A1g+B1g+E uA1g B1g E us d x2-y2(p x、p y)d z2两种类型:dsp2(d x2-y2、s、p x、p y)、d2p2(d z2、d x2-y2、p x、p y)5)八面体AB6(O h) 例:[Fe(H2O)6]3+O h E 8C3 6C26C4 3C2i 6S4′8S6 3σh 6σdΓ 6 0 0 2 2 0 0 0 4 2约化得:Γ=A1g+E g+T1u A1g E gT1us (d z2、d x2-y2) (p x、p y、p z)只有唯一的d2sp3杂化(d z2、d x2-y2、s、p x、p y、p z)3、π成键杂化方案在AB n分子中,原子A上要有2n个π型杂化轨道和在B原子上的2n个π原子轨道成键。


第二章配合物的化学键理论内容:研究中心原子和配体之间结合力的本性。
目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出一、理论要点:①配体的孤对电子可以进入中心原子的空轨道;中心原子总是用空轨道杂化,然后用杂化轨道接收配体提供的孤对电子。
②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
中心原子的价层电子结构与配体的种类和数目共同决定杂化类型。
③杂化类型决定配合物的空间构型,磁距和相对稳定性。
二、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-sp杂化直线型[AgCl2]-三、内轨型和外轨型若要形成ML6型配合物(L为单齿配体),则需要6个空杂化轨道接收6个L提供的孤电子对,满足该条件的杂化类型有d2sp3和sp3 d2。
尽管这两种杂化都导致八面体型配合物,但前者是次外层(n-1)d轨道,而后者是最外层nd轨道,因此与这两种杂化相应的配合物分别称内轨型和外轨型配合物。
中心原子的价层电子数和配体的性质都是影响配合物内轨型和外轨型的因素。
当d电子数≤3时,该层空d轨道≥2,总是生成内轨型配合物。
当中心原子价层d电子数为7~10时,即使强制d轨道中的电子配对,所能得到的该层空d轨道数也小于2,因此只能用最外层d轨道参与杂化,总是生成外轨型配合物。
当中心原子价层d电子数为4~6时,对于配位能力较强的配体,即配位原子电负性较小,容易给出孤电子对,对中心原子价层d电子排布影响较大,强制d电子配对,空出2个价层d轨道参与d2sp3杂化,生成内轨型配合物.若配体的配位能力较弱,即配位原子电负性较大,则不易给出孤电子对,对中心原子价层d电子排布影响较小,只能用最外层d轨道参与杂化,生成外轨型配合物。
配位场理论ligand field theory说明和解释配位化合物的结构和性能重要的理论有价键理论、晶体场理论、分子轨道理论和配位场理论。
配位化合物的价键理论根据配位化合物的性质,按杂化轨道理论用共价配键和电价配键解释配位化合物中金属离子和配位体间的结合力。
例如呈现反磁性,是由于中心离子有未充满的d轨道和s,p空轨道,这些空轨道通过杂化组成杂化轨道,由配位体提供孤对电子;配位体L与中心离子M之间形成L→M的σ键。
是顺磁性的。
中心离子的未成对电子数目和自由离子一样,认为金属离子和配位体以静电吸引力结合在一起。
价键理论简明地解释配位化合物的几何构型和配位化合物的磁性等性质。
价键理论没有提到反键轨道,不能满意解释配位化合物的光谱数据。
晶体场理论是静电作用模型。
把中心离子(M)和配位体(L)的相互作用看作类似离子晶体中正负离子的静电作用。
当L接近M时,M中的d轨道受到L负电荷的静电微扰作用,使原来能级简并的d轨道发生分裂。
按微扰理论可计算分裂能的大小,因计算较繁,定性地将配位体看作按一定对称性排布的点电荷与M的d轨道电子云产生排斥作用。
由于d轨道分布的特点,在配位场中原来5个能级简并的d轨道能级发生分裂,引起电子排布及其他一系列性质的变化,比如电子将重新分布,体系能量会降低,据此解释配位化合物的各种性质。
例如八面体配位离子中,d轨道分裂成两组:低能级的dxy,dxz,dyz,它们三者的能量相等,称为t2g(2g为下标)轨道,此二者的能量相等;高能级的dx2-y2d,dz2,称为eg(g为下标)轨道。
这两组能级间差值称为晶体场分裂能Δ ,配体场强越大,分裂能值越大。
d电子根据Δ和成对能(P)相对大小填在这两组轨道上,形成强场低自旋和弱场高自旋结构。
在不同构型的配合物中,中心离子d轨道能级分裂情况不同。
以此成功地解释了配位化合物的结构、光谱、稳定性及磁性等一系列性质。
配位化合物的分子轨道理论是用分子轨道理论的观点和方法处理金属离子和配位体成键作用。
配合物的化学键理论The Chemical Bond Theories of Complexes配合物的化学键理论处理中心原子(或离子)与配体之间的键合本质问题,用以阐明中心原子的配位数、配位化合物的立体结构以及配合物的热力学性质、动力学性质、光谱性质和磁性质等。
几十年来,提出来的化学键理论有: 静电理论(EST) Electrostatic Theory 价键理论(VBT) Valence Bond Theory 晶体场理论(CFT) Crystal Field Theory分子轨道理论(MOT) Molecular Orbital Theory 角重叠模型(AOM) Angular Overlap Model在这一节中,我们讲授配合物的价键理论和晶体场理论。
分子轨道理论和角重叠模型在后续课程中学习。
一、价键理论(Valence Bond Theory )L .Pauling 等人在二十世纪30年代初提出了杂化轨道理论,首先用此理论来处理配合物的形成、配合物的几何构型、配合物的磁性等问题,建立了配合物的价键理论,在配合物的化学键理论的领域内占统治地位达二十多年之久。
1.价键理论的基本内容:(1) 配合物的中心体M 与配体L 之间的结合,一般是靠配体单方面提供孤对电子对与M 共用,形成配键M ←∶L ,这种键的本质是共价性质的,称为σ配键。
(2) 形成配位键的必要条件是:配体L 至少含有一对孤对电子对,而中心体M必须有空的价轨道。
(3) 在形成配合物(或配离子)时,中心体所提供的空轨道(s 、p ,d 、s 、p 或s 、p 、d)必须首先进行杂化,形成能量相同的与配位原子数目相等的新的杂化轨道。
2.实例:(1) 主族元素配合物 Be 4O(CH 3COO)6:每个Be 原子都采取sp 3杂化-4BF :B 原子为sp 3杂化,正四面体构型 -36AlF :-3][ Al 3+周围共有12个价电子 Al 3+采取sp 3d 2杂化 (2) 过渡元素配合物a .(n - 1)d 10电子构型中心体+243)Zn(NH sp 3杂化 正四面体-3HgI sp 2杂化 平面三角形b .(n - 1)d 8电子构型中心体F Al F F F FF+243])[Ni(NH sp 3杂化 正四面体 -24]Ni(CN)[ dsp 2杂化 平面四方-24PtCl dsp 2杂化 平面四方c .(n - 1)d x (x <8)电子构型中心体-36Fe(CN) d 2sp 3杂化 正八面体+363])[Co(NH d 2sp 3杂化 正八面体 +263])[Co(NH sp 3d 2杂化 正八面体-36FeF sp 3d 2杂化 正八面体3.讨论:(1) 配合物中的中心体可以使用两种杂化形式来形成共价键:一种杂化形式为(n - 1)d 、n s 、n p 杂化,称为内轨型杂化。
第二章配合物的化学键理论内容:研究中心原子和配体之间结合力的本性。
目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出一、理论要点:①配体的孤对电子可以进入中心原子的空轨道;中心原子总是用空轨道杂化,然后用杂化轨道接收配体提供的孤对电子。
②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
中心原子的价层电子结构与配体的种类和数目共同决定杂化类型。
③杂化类型决定配合物的空间构型,磁距和相对稳定性。
二、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-sp杂化直线型[AgCl2]-三、内轨型和外轨型若要形成ML6型配合物(L为单齿配体),则需要6个空杂化轨道接收6个L提供的孤电子对,满足该条件的杂化类型有d2sp3和sp3 d2。
尽管这两种杂化都导致八面体型配合物,但前者是次外层(n-1)d轨道,而后者是最外层nd轨道,因此与这两种杂化相应的配合物分别称内轨型和外轨型配合物。
中心原子的价层电子数和配体的性质都是影响配合物内轨型和外轨型的因素。
当d电子数≤3时,该层空d轨道≥2,总是生成内轨型配合物。
当中心原子价层d电子数为7~10时,即使强制d轨道中的电子配对,所能得到的该层空d轨道数也小于2,因此只能用最外层d轨道参与杂化,总是生成外轨型配合物。
当中心原子价层d电子数为4~6时,对于配位能力较强的配体,即配位原子电负性较小,容易给出孤电子对,对中心原子价层d电子排布影响较大,强制d电子配对,空出2个价层d轨道参与d2sp3杂化,生成内轨型配合物.若配体的配位能力较弱,即配位原子电负性较大,则不易给出孤电子对,对中心原子价层d电子排布影响较小,只能用最外层d轨道参与杂化,生成外轨型配合物。
类似地,对于ML4型配合物,(L 为单齿配体)当中心原子价层d电子数为5~8时,若配体较强,则dsp2杂化,生成内轨型平面四方形配合物。
例如[Ni(CN)4]2-。
若配体较弱,则sp3杂化,生成外轨型四面体型配合物,例如[Ni(NH3)4]2+。
四、用价键理论说明或判断配合物的性质1)配合物磁性与配合物中成单电子数的关系理论分析表明:配合物的分子磁矩μ与配合物中未成对电子数n 有关。
如:对某些配合物:µ=[n(n+2)]1/2 B.M.1B.M. = 9.27×10-21erg·G-12)实验发现:同种金属离子的不同配合物有时具有不同的磁矩。
如:K4[Fe(CN)6] µ=0.00 B.M.FeSO4.7H2Oµ=4.90 B.M.这表明,这两种配合物中成单电子数不同。
3) 价键理论的解释(内、外轨型配合物)内轨型配合物,如:K4[Fe(CN)6 ]自由Fe2+( d 6 ):重排为:[Fe(CN)6]4-配合物利用内层的(n-1)d轨道形成杂化成键轨道,因此称为内轨型配合物。
成单电子数较自由Fe2+下降。
外轨型配合物:如[Fe (H2O)6]2+利用nd轨道杂化成键,称为外轨型配合物。
习题:根据实验测得有效磁矩,判断下列各配离子是低自旋还是高自旋,是内轨型还是外轨型,中心离子杂化类型,配离子的空间构型。
(1) [Fe(en)3]2+ 5.5 B.M(2) [Co(SCN)4]2- 4.3 B.M(3) [Mn(CN)6]4- 1.8 B.M(4) [FeF6]3- 5.9 B.M(5) [Ni(CN)4]2-0 B.M(6) [Ni(NH3)4]2+ 3.2 B.M五、价键理论的成功与不足1)成功简单明了,容易理解,能说明配位数、空间构型、磁性、稳定性等。
2)不足①不能解释d能级的分裂。
②在形成配键时,3d和4d轨道的利用有任意性。
③不能解释配合物的电子光谱。
④不能解释和预测磁性的细节。
⑤定量程度差,只是一种近似的定性理论。
而无法解释配合物的吸收光谱。
⑥无法说明Cu2+平面正方形内轨型配合物的稳定性如[Cu(NH3)4]2+:据此,4p电子很容易失去,但事实上Cu2+不易被氧化练习:实验测得下列化合物中,⑶、⑷是高自旋物质。
试根据价键理论绘出这些配离子的杂化轨道图,它们是内轨型,还是外轨型配合物:⑴[Ag(NH3)2]+⑵[Zn(NH3)4]2+⑶[CoF6]3–⑷[MnF6]4–⑸[Mn(CN)6]4−解:⑴Ag+ 的价层电子构型为4d10,所以[Ag(NH3)2]+ 配离子中,中心原子Ag+ 只能采取sp杂化,形成外轨型配合物。
[Ag(NH3)2]+ 配离子中Ag+ 的价层电子排布为4dsp 5p[Ag(NH3)2]+⑵Zn2+ 的价层电子构型为3d10,所以[Zn(NH3)4]2+ 配离子中,中心原子Zn2+ 只能采取sp3杂化,形成外轨型配合物。
[Zn(NH3)4]2+ 配离子中Zn2+ 的价层电子排布为3dsp3[Zn(NH3)4]2+⑶ Co3+ 的价层电子构型为3d6。
中心原子的这种价层d电子构型既可形成外轨、高自旋的配合物,又可形成内轨、低自旋的配合物。
究竟形成哪种类型的配合物,这取决于配体的性质。
实验测得[CoF6]3−配离子为高自旋的,所以中心原子Co3+ 采取sp3d2杂化,形成外轨型的的配合物:3dsp3d2 4d[CoF6]3−⑷Mn2+ 的价层电子构型为3d5。
同样这种价层d电子构型的中心原子,视配体的不同可以形成不同类型的配合物。
实验测得[MnF6]4−配离子为高自旋的,所以中心原子Co3+ 采取sp2d2杂化,形成外轨型的的配合物:3dsp3d2 4d[MnF6]4−⑸Mn2+的价层电子构型为3d5,而且CN−为强场配体,所以[Mn(CN)6]4−配离子中,中心原子Mn2+ 只能采取d2sp3杂化,形成内轨型配合物:3dd2sp3[Mn(CN)6]4−第二节晶体场理论(Crystal field theory)一、概述由Bethe和Van Vleck提出这一理论在静电理论的基础上把中心离子和配位体看成是点电荷或偶极子,配合物的成键由等带正电荷的中心离子和带负电荷的配体之间静电吸引形成,配体间相互排斥,配位体电子对中心离子最外层电子(特别是过渡金属元素离子的d电子)有排斥作用.1.基本要点:①在配合物中,中心离子M处于带电的配位体L形成的静电场,二者完全靠静电作用结合在一起;②晶体场对M的d电子产生排斥作用,使M的d电子轨道发生能级分裂;③分裂类型与化合物的空间构型有关;④晶体场相同,L不同,分裂程度也不同.* 配位场理论:若不是单纯的采用静电模型,而是允许金属离子与配体间存在某种共价键,则这种经过修改的晶体场理论称为配位场理论。
* 记注:晶体场理论和配位场理论只是把注意力集中到配合物总生成能中一个小的方面,即CFSE。
二、d 轨道能级分裂(单电子能级的分裂)1、定义:由于d 轨道空间取向不同,与非球形对称静电场的作用则不相同,引起d 轨道能级发生分裂。
2、正八面体场中d 轨道的分裂1)d 轨道与电场的作用xxyd z 2 d x 2-y 2 d xy d yz d xz2)能级计算:E=Dq2_y2 d (d xy,d yz,, o10自由离子d轨道球形场八面体场e g与t2g能量差,称为分裂能Δo=E eg-E t2g=10Dq (1)能量重心原理:量子化学证明,球形场能级分裂后,5个d轨道总能量应保持不变,即2E eg+3E t2g=5Es。
若取Es为能量零点,则2E eg+3E t2g=0 (2)联合(1)与(2)方程,解得Eeg=6DqE t2g=-4Dq即能级分裂后,e g轨道上升6Dq,t2g轨道能量下降4Dq。
3、正四面体场中d轨道能级的分裂1) d轨道与电场的作用d x2-y2d xy极大值指向面心极大值指向棱的中点2)能级计算:E=10Dq(d xy, d yz, d xz)49tx2-y2, dz2)自由离子球形场四面体场配体相同,中心离子与配体距离相同时,分裂能Δt=4/9Δo即Δt = E t2 - E e = 4/9Δo Dq---------(1)同理,若选Es为能量零点, 则3E t2+2E e=5E s=0---------(2)联立(1)和(2),解出:E t2=1.78Dq, E e=-2.67Dq能级分裂结果;t2轨道能量上升1.78Dq,e轨道能量下降2.67Dq。
4、各种对称性场中d轨道能级分裂后的能量(略)三、d轨道中电子的排布及对配合物磁性的解释1、分裂能与成对能:分裂能:当一个电子由低能的d 轨道进入高能d轨道时所需要的能量(Δ)。
成对能:迫使本来自旋平行分占两个轨道的电子挤到同一轨道,所需的能量(P)。
强场:如果这两种作用中,配体场的作用较大,而电子相互作用较小,这称为强场情况。
Δ>P(强场)弱场:如果这两种作用中,电子相互作用较大,而配体场的影响较小,这称为弱场情况。
Δ<P(弱场)2.d轨道中电子的排布规则:与Δ和P的相对大小有关Δ<P(弱场)时,按高自旋排布;Δ>P(强场)时,按低自旋排布。
3、八面体配合物* 不论强、弱场,d1、d2、d3、d8、d9、d10电子排布相同,无高、低自旋之分。
** d4、d5、d6、d7有高、低自旋之分。
即可解释磁性,如K4[Fe(CN)6] µ=0.00 B.MFeSO4.7H2Oµ=5.10 B.M第一过渡系d4~d7离子的Δo值与P值的比较4、四面体配合物由于相同情况下,Δt=4/9Δo, 因此一般情况下,Δ<P。
即在四面体配合物中,大多为弱场情况,采取高自旋排布。
练习:Cr2+、Cr3+、Mn2+、Fe2+、Fe3+、Co2+、Co3+ 离子在八面体强场和弱场中各有多少单电子,绘图说明之。
解答:四、晶体场稳定化能(CFSE)1、定义:当d电子从未分裂的球形场d轨道Es能级进入分裂的d轨道所产生的总能量下降值, 称为CFSE。
2、CFSE的计算1)八面体场的CFSEE s = 0e g 6Dqt2g -4Dqt2g轨道上进入1个电子能量下降4Dqe g轨道上进入1个电子能量上升6DqCFSE=E S– E晶体场=0- E晶体场例:d6组态金属离子,在弱八面体场中, 电子排布为(t2g)4(e g)2CFSE=0-【4×(-4Dq)+2×6Dq】=4Dq而在强八面体场中, d电子排布为低自旋(t2g)6(e g)0CFSE=0-[6×(-4Dq)+2P]=24Dq -2P* 低自旋时,要注意成对能P。