价键理论
- 格式:doc
- 大小:81.00 KB
- 文档页数:2

价键理论自1916年路易斯提出经典的共价键理论以来,共价键理论有了很大的发展。
现代共价键理论有两种,一是价键理论,二是分子轨道理论。
(一)价键理论的基本要点价键理论,又称电子配对法,其基本要点如下:1.具有自旋相反的未成对电子的两个原子相互接近,可以形成稳定的共价键。
如果A、B两个原子各有一个自旋相反的未成对的电子,那么这两个未成对电子可以相互配对形成稳定的共价键,这对电子为A、B两原子所共有(共用)。
如果A、B各有两个或三个未成对的电子,则自旋相反的单电子可两两配对形成双键或叁键。
如果A原子有两个未成对电子,B原子有一个未成对电子,那么一个A原子能与两个B原子结合形成AB2型分子,…。
2.原子中未成对的电子数等于原子所能形成的共价键数目(共价键的饱和性)。
共价键是由成键原子中自旋相反的未成对电子配对形成的。
一个原子的一个电子和另一个原子的一个电子配对以后,不能再和第二个电子配对。
因为这时其中必有两个电子的自旋方向相同而相斥。
也就是说一个原子所能形成共价键的数目是一定的。
原子中未成对的电子数等于原子所能形成的共价键数目,这就是共键价的饱和性。
例如,H原子只有一个未成对电子,它和另一个H原子的未成对电子配对后,就不能再与第二个H原子的电子配对了,……。
3.成键电子的电子云重叠越多,核间电子子云密度就越大,形成的共价键就越牢固(共价健的方向性)。
共价键的生成是由于自旋相反的单电子相互配对,电子云重叠的结果。
因此,当两个原子形成分子时,电子云重叠的程度越大,则两原子间的电子云密度越大,生成的共价键就越牢固,所以,在形成共价键时,电子云总是尽可能达到最大程度的重叠。
因此,在形成共价键时,原子间总是尽可能沿着电子云最大重叠方向成键。
s电子云呈球形对称分布,p、d、f电子云在空间都有一定的伸展方向。
在形成共价键时,除了s 电子云和s电子云可以在任何方向上都能达到最大程度的重叠外,p、d电子云的重叠,只有在一定方向上才能使电子云有最大程度的重叠。

价键理论概述价键理论概述摘要:价键理论是指固体或分⼦中原⼦的价电⼦结构和原⼦与原⼦之间形成的键以及两者关系的理论。
它是从原⼦和原⼦结构层次, 深⼊了解材料⼀种重要理论, 能帮助⼈们设计满⾜需要的新材料。
根据收集到的资料, 对价键理论及其应⽤进⾏扼要地归纳与阐述。
关键词:价键理论共价键键参数⾦属应⽤价键理论起源于1916 年美国科学家G1 N1Lew is[1]提出的电⼦配对理论。
1927 年德国科学家W1 He itler与F1 L London[2]第⼀个⽤量⼦⼒学处理H2分⼦, 揭⽰了共价键的本质。
1930 年前后Pauling[3]和S later[4]等把这个理论发展成为⼀种全⾯的键理论, 称为价键理论。
⾦属的价键理论实质就是⽤电⼦配对法来处理⾦属键。
这⼀理论在⾦属材料中有着重要的指导作⽤, 它能帮助⼈们从电⼦结构和原⼦结构层次了解晶体结构, 并以此寻找需要的⾦属新材料。
因此, 国内外科学家, 在这⽅⾯做了⼤量的⼯作, 鉴于价键理论的重要性, 对其发展与应⽤做扼要的归纳与阐述。
⼀、键价理论的基本知识1.基本概念价键理论是在Pauling 离⼦晶体电价规则基础上发展起来的, 它继承了电价规则中/原⼦的价分配在原⼦所连诸键上0的基本概念, 同时允许原⼦所连诸键的键价做不均匀的分配。
价鍵的主要内容包括以下⼏个⽅⾯:(1)在价键理论或价键法则中, 将在反应中保持不变的最基本的实体称作原⼦。
在由⼴义( Lewis)酸(阳离⼦)与⼴义碱(阴离⼦)组成的离⼦性化合物中, 荷正电者为正价, 荷负电者为负价。
(2)化学计量要求离⼦性(或酸碱)化合物中的总正价与总负价的绝对值相等。
即化合物整体保持电中性的原理。
(3)原⼦以化学键与其近邻原⼦键合, 其键连原⼦数称为该原⼦的配位数, 此数亦为该原⼦参与化学键的成键数。
(4)价键理论认为, 原⼦的价将分配在它所参与的诸键上, 使每个键均有⼀定的键价, 并符合价和规则。





价键理论价键理论valence-bond theory,一种获得分子薛定谔方程近似解的处理方法。
又称电子配对法。
历史上最早发展起来的化学键理论。
主要描述分子中的共价键和共价结合,其核心思想是电子配对形成定域化学键。
1产生1927年W.H.海特勒和F.W.伦敦首次完成了氢分子中电子对键的量子力学近似处理,这是近代价键理论的基础。
L.C.鲍林等加以发展,引入杂化轨道概念,综合成价键理论,成功地应用于双原子分子和多原子分子的结构。
价键理论与化学家所熟悉的经典电子对键概念相吻合,一出现就得到迅速发展。
但价键理论计算比较复杂,使得后来发展缓慢。
随着计算技术日益提高,该理论还会有新发展。
1927年,Heitler 和London 用量子力学处理氢气分子H2,解决了两个氢原子之间化学键的本质问题,使共价键理论从典型的Lewis理论发展到今天的现代共价键理论。
海特勒-伦敦方法处理氢分子氢分子的哈密顿算符是:式中rA1、rB1为核A、B与电子1之间的距离;r12为两个电子之间的距离;RAB为两个原子核之间的距离……(图1);1/RAB表示两个原子核之间的势能(氢核和电子电荷皆为1基本电荷单位);1/rA1、1/rB1、…也是势能;墷是拉普拉斯算符。
海特勒-伦敦方法的要点在于如何恰当地选取基态H2的近似波函数Ψ(1,2)(或称尝试波函数),然后用变分公式使氢分子能量E为最低(假定Ψ是归一化的):式中*表示复数共轭。
考虑两个氢原子组成的体系,若两个氢原子A(有电子1)和B(有电子2)的基态波函数为:φA⑴=πexp(-rA1)φB⑵=πexp(-rB2)假如两个氢原子相距很远,那么体系波函数是:Φ1(1,2)=φA⑴φB⑵实际上两个电子是不可区分的。
同样合适的函数是:Φ2(1,2)=φB⑴φA⑵两个函数Φ1和Φ2都对应相同的能量。
海特勒和伦敦就取两个函数的等权线性组合作为H2的变分函数:Ψ(1,2)=c1Φ1+c2Φ2解久期方程得c1=±c2,波函数和能量是:式中s称原子轨道的重叠积分。



第六章 价键理论价键理论,顾名思义:就是有关分子间化学键的理论。
其核心思想是电子两两配对形成定域的化学键,因而价键理论又称为电子配对理论。
价键理论是在Heitler-London 处理2H 问题的基础上发展起来的。
因此,我们先来回顾一下Heitler-London 方法。
§6.1 Heitler-London 方法在讨论2H 时,H.L 从Pauli 原理出发,即电子轨道自旋波函数必须是反对称的,直接推出了2H 的单态和三态波函数,)()()()())1,31212ab ba ψαββα=± ab ba +:对称的 αββα-:反称 ab ba -:反称 αββα+:对称∴两两组合,只能有+→-⎛⎫⎪-→+⎝⎭a ,b 为归一化的实函数,为氢原子的原子轨道,a 表示在A 核的b 表示在B 核的 |a b S =体系能量(相应于单态和三态):1,31,31,3ˆE Hψψ=∵ˆH不含自旋,∴ψ中自旋可积去,为1,剩下E 只是空间轨道在ˆH 下的作用结果。
∴()1,321ˆ21E ab ba H ab ba S =±±+ ()121ˆ21E ab ba Hab ba S =++⨯+()()21ˆˆˆˆ21ab Hab ab H ba ba H ab ba H ba S =++++ ∵ˆˆab H ab ba H ba =;ˆˆab H ba H ab = ()()21ˆˆ1ab H ab ab H ba S =++ 同理:()()21ˆˆ1ab H ab ab H ba S =-- 写在一起:()()1,321ˆˆ1E ab H ab ab H ba S =±± 定义ˆQ a b H a b = 库仑作用能ˆK ab Hba = 交换作用能 2H 分子在Hamiltonian 在定核近似下,(只与电子坐标有关)可写为: ()()121ˆˆˆ12Hh h r =++ ∴()1,3211E Q K S=±± 其中 Q ab H ab a h a b h b ab g ab ==++ ∵a ,b 对称(a 为A 核1s ,b 为B 核1s ,A ,B 核一样), ∴ 2Q a h a a b g a b=+()()()()()11122K a b H b a a h b b a b h a Sa b g a b==++ S 2S a h b ab g ab =+∵两归一化在函数内积总是小于等于1,即|1a b s =≤"1"=只有 a b = 时∴在我们这种情况下,21S <作为一种近似我们可以把21S +的2S 省去,则能量表示变为,1,3E Q K =± <0 >0∵0K < (2K S a h b ab g ab =+)这一项积分总很小 ∴13E E <即单态为基态,较稳定,符合事实,另外,我们不能省去K 中的S ,如果省去,则K>0,从而31E E <,就不对了。
配位场理论ligand field theory说明和解释配位化合物的结构和性能重要的理论有价键理论、晶体场理论、分子轨道理论和配位场理论。
配位化合物的价键理论根据配位化合物的性质,按杂化轨道理论用共价配键和电价配键解释配位化合物中金属离子和配位体间的结合力。
例如呈现反磁性,是由于中心离子有未充满的d轨道和s,p空轨道,这些空轨道通过杂化组成杂化轨道,由配位体提供孤对电子;配位体L与中心离子M之间形成L→M的σ键。
是顺磁性的。
中心离子的未成对电子数目和自由离子一样,认为金属离子和配位体以静电吸引力结合在一起。
价键理论简明地解释配位化合物的几何构型和配位化合物的磁性等性质。
价键理论没有提到反键轨道,不能满意解释配位化合物的光谱数据。
晶体场理论是静电作用模型。
把中心离子(M)和配位体(L)的相互作用看作类似离子晶体中正负离子的静电作用。
当L接近M时,M中的d轨道受到L负电荷的静电微扰作用,使原来能级简并的d轨道发生分裂。
按微扰理论可计算分裂能的大小,因计算较繁,定性地将配位体看作按一定对称性排布的点电荷与M的d轨道电子云产生排斥作用。
由于d轨道分布的特点,在配位场中原来5个能级简并的d轨道能级发生分裂,引起电子排布及其他一系列性质的变化,比如电子将重新分布,体系能量会降低,据此解释配位化合物的各种性质。
例如八面体配位离子中,d轨道分裂成两组:低能级的dxy,dxz,dyz,它们三者的能量相等,称为t2g(2g为下标)轨道,此二者的能量相等;高能级的dx2-y2d,dz2,称为eg(g为下标)轨道。
这两组能级间差值称为晶体场分裂能Δ ,配体场强越大,分裂能值越大。
d电子根据Δ和成对能(P)相对大小填在这两组轨道上,形成强场低自旋和弱场高自旋结构。
在不同构型的配合物中,中心离子d轨道能级分裂情况不同。
以此成功地解释了配位化合物的结构、光谱、稳定性及磁性等一系列性质。
配位化合物的分子轨道理论是用分子轨道理论的观点和方法处理金属离子和配位体成键作用。
第三章配合物的化学键理论内容:研究中心原子和配体之间结合力的本性。
目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
四种理论:①价键理论、②晶体场理论、③分子轨道理论、④角重叠模型第一节价键理论(Valence bond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-sp杂化直线型[AgCl2]-二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)轨道波函数是与轨道符号下标多项式按相同的方式变换的。
*在注意到特征标表右边某列中轨道的多项式标记后,即可确定轨道的变换性质。
*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A2″:p zE″:(d xz、d yz)2、σ轨道杂化方案(如何确定某一组杂化轨道由哪些原子轨道组成)1)四面体分子AB4(Td)[CoCl4]2-原子A以哪些原子轨道组成在原子A上四个σ轨道的集合,其中每个轨道的瓣指向B原子。
以四个杂化轨道的集合(或四个B原子上指向A 原子的σ轨道的集合)作为分子点群(Td)表示的基,确定该表示的特征标:①不变(1)对杂化轨道波函数的操作有三种情况:②改变符号(-1)③与其他函数变换(0)r1r2r3r4恒等操作,χ(E)=4 C3操作,χ(C3)=1对C2、S4和σd用同样方法处理,得T d E 8C3 3C26S46σdΓ 4 1 0 0 2约化:T d E 8C3 3C2 6S46σdA1 1 1 1 1 1A2 1 1 1 -1 - 1E 2 -1 2 0 0 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -1 1 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+6×(-1)×2]=0a(E)= 1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+6×0×2]=0 a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24[3×4+8×0×1+3×(-1)×0+6×(-1)×0+6×1×2]=1约化结果Γ=A1+T2这说明组成杂化轨道的四个原子轨道中,必有一个是属于A1表示的原子轨道,另三个为属于T2表示的三个原子轨道。
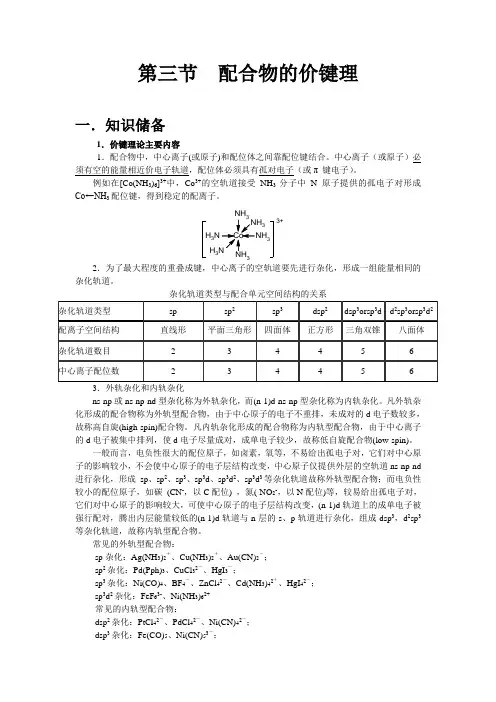
第三章配合物结构第二节配合物的化学键理论3.2.1 价键理论1930年代,由L. Pauling提出。
1.价键理论的要点:(1) 形成体(M):有空轨道;配位体(L):有孤对电子;二者形成配位键M L(2) 形成体(中心离子)采用杂化轨道成键(3) 杂化方式与空间构型有关配位数杂化类型空间构型2 sp 直线3 sp2平面三角形4sp3正四面体4dsp2正方形5 sp3d 三角双锥体6 sp3d2/d2sp3 正八面体2.配位数为 2 的配合物[Ag(NH3)2]+的空间构型为直线形,μ=0。
此外:[AgCl]-,[CuCl2]-2[BeX 4]2-的空间构型为四面体。
3.配位数为 4 的配合物 [Ni(CN)4]2-的空间构型为平面正方形,μ=0这类配合物绝大多数是八面体构型,形成体可能采取d2sp3或sp3d2杂化轨道成键。
4.配位数为6 的配合物例如:[Fe(CN)6]3- ,μ=2.4B.M. ;内轨配键。
以内轨配键形成的配合物叫内轨型配合物。
例如:[FeF6]3- ,μ=5.90B.M.外轨配键。
以外轨配键形成的配合物叫外轨型配合物。
同一中心离子的内轨型配合物比外轨型配合物稳定。
([Fe(CN)6]3-) =52.6, ([FeF 6]3-) = 14.3 lg lg 外轨型配合物: [FeF 6]3-(1) 分子几何构型:正八面体(2) 稳定性:外轨型配合物稳定性低. (3) 磁性: 顺磁性3d 5 : 3d 1xy 3d 1xz 3d 1yz 3d 1x2-y2 3d 1z2 成单电子数 n = 5,磁矩μm =B.M. 高自旋(High spin )配合物)2(+n n 内轨型配合物 Fe(CN)63-①分子几何构型为正八面体②稳定性: (n -1)d 2nsnp 3杂化 → 内轨型配合物,稳定性高 ③磁性: 成单电子数为1,顺磁性↓, 3d 5 : 3d 1xy 3d 2xz 3d 2yz (3d x2-y20 3d z2 0参加杂化)低自旋(Low spin )配合物价键理论的优缺点:• 很好地解释了配合物的空间构型、磁性、稳定性,直观明了。
化学键的价键理论共价键价电子对化学键是化学反应中最基本的概念之一,它描述了原子之间的结合方式。
化学键的形成涉及到共享或转移电子,其中共价键是最常见的一种化学键类型。
共价键形成的基本单位是价电子对,本文将探讨化学键的价键理论及共价键价电子对的性质。
1. 共价键的概念共价键是原子之间通过电子的共享形成的化学键。
在共享过程中,原子通过共享价电子对形成共价键,使得原子能量降低并达到更稳定的状态。
共价键可以形成在同种元素之间(如氢气分子H2)或不同元素之间(如氧气分子O2)。
2. 价键理论2.1 原子轨道和价电子对根据价键理论,原子由核心和围绕核心的电子组成。
电子存在于不同的轨道中,其中价电子是参与形成化学键的电子。
价电子对是共价键的基本单位,可以是一个或多个共享的电子对。
2.2 原子轨道的杂化原子的轨道通过杂化可以重新组合成新的轨道,以适应共享电子对的形成。
常见的杂化类型包括sp、sp2和sp3杂化。
sp杂化产生线性共价键,sp2杂化产生三角平面共价键,sp3杂化产生四面体共价键。
2.3 共价键的形成共价键的形成通过轨道重叠实现。
共价键的形成有两种基本的重叠方式:头-头重叠和边-边重叠。
在共价键形成中,电子云发生重叠,形成化学键。
3. 共价键价电子对的性质3.1 共价键的长度和键能共价键的长度取决于原子核间的距离,较短的键对应于较强的共价键。
共价键的键能是破坏化学键所需的能量,与键的强度相关。
3.2 共价键的极性共价键可以是非极性的或极性的。
非极性共价键是由相同或相似元素之间的共享电子对形成的,如氢气分子H2。
极性共价键是由不同元素之间的共享电子对形成的,电子云偏向电负性较高的原子。
3.3 共价键的结构和分子形状共价键决定了分子的结构和形状。
共价键的方向性以及原子杂化方式对分子形状产生重要影响。
杂化sp3形成四面体分子结构,sp2杂化形成平面三角形分子结构。
4. 应用和意义共价键的理论对于解释化学反应、分子形状及性质具有重要意义。