无机化学第十八章-配位化合物的价键理论
- 格式:docx
- 大小:258.16 KB
- 文档页数:29

配合物的价键理论配合物中的化学键主要是指配合物内中心离子(或原子)M 与配体L 之间的化学键。
中心离子和配体之间通过什么样的作用力结合在一起?这种结合力的本质是什么?为什么配离子具有一定的空间构型而稳定性又各不相同?19世纪末,维尔纳(Werner A )曾试图回答这些问题,但没有成功。
直到20世纪,在近代原子和分子结构理论建立以后,用现代的价键理论以及晶体场理论、配位场理论和分子轨道理论,才较好地阐明了配合物中化学键的本质。
1931年鲍林首先将分子结构的价键理论应用于配合物,后经他人修正补充,逐步完善成配合物的现代价键理论。
1.配合物价键理论的要点(1) 中心离子(或原子)M 与配体L 形成配合物时,中心离子(或原子)以空的价轨道接受配体中配位原子提供的孤对电子,形成σ配键(用M ←L 表示)。
(2) 中心离子(或原子)所提供的空价轨道必须杂化,与配位原子的充满孤对电子的原子轨道相互重叠,形成配位共价键。
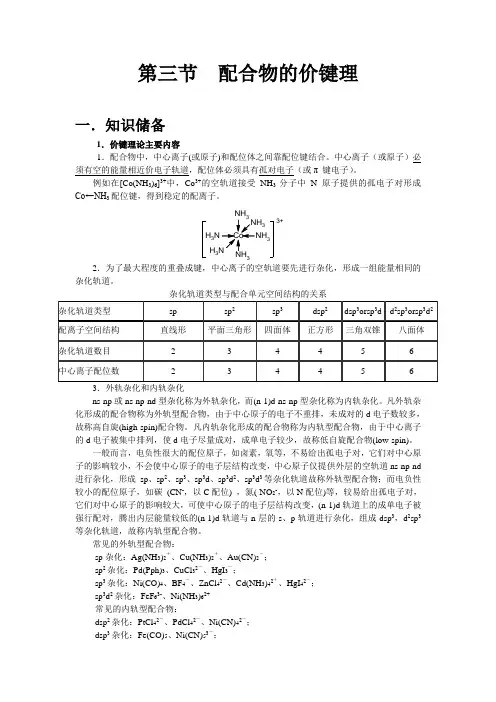
2.中心离子轨道杂化的类型 在配合物的形成过程中,中心离子需提供一定数目的经杂化的能量相同的空的价轨道与配体形成配位键。
中心离子所提供的空轨道的数目,由中心离子的配位数所决定,故中心离子空轨道的杂化类型与配位数有关。
中心离子空轨道的杂化类型除了前面讲过的sp 、sp 2、sp 3杂化外,能量相近的(n-1)d ,n d 轨道也能参与杂化。
(1) 配位数为2的中心离子的杂化类型 讨论[Ag(NH 3)2]+ 配离子的形成 Ag +离子的价电子层结构为: Ag +离子和NH 3形成[Ag(NH 3)2]+配离子时,配位数为2,Ag +需提供二个空轨道。
Ag +离子外层能级相近的一个5s 和一个5p 轨道经杂化,形成二个等价的sp 杂化轨道,容纳二个NH 3中二个配位N 原子提供的二对孤对电子,形成二个配键(虚线内杂化轨道中的共用电子对由配位氮原子提供): 两个sp 杂化轨道在空间成180°,故[Ag(NH 3)2]+配离子的空间构型呈直线形。

无机化学中的配位化合物无机配位化合物是指由中心金属离子或原子与周围配体形成的稳定化合物,其中配体可以是有机分子、无机物以及某些复杂的大分子。
这些化合物在化学、材料和生物领域具有广泛的应用。
本文将对无机化学中的配位化合物进行详细介绍。
一、配位键的形成在配位化合物中,中心金属离子通过与配体的配位键结合在一起。
配位键可以是共价键,也可以是离子键。
在共价配位键中,金属离子与配体共享电子对,形成共有的化学键。
而离子配位键中,金属离子通过吸引配体上的电子形成离子键。
二、常见的配体在配位化合物中,各种不同的配体可以与中心金属离子形成配位键。
常见的配体包括一价的阴离子(如Cl-、Br-、I-)、二价的阴离子(如O2-、OH-)以及有机分子(如NH3、CO、CN-等)。
这些配体的不同基团和电性决定了它们与金属离子之间的相互作用方式和配位键的强度。
三、配位化合物的结构配位化合物的结构可以是简单的一对一结构,也可以是复杂的多中心配位结构。
在一对一结构中,一个中心金属离子配位于一个配体上。
而在多中心配位结构中,一个或多个中心金属离子与多个配体形成配合物。
四、配位化合物的性质配位化合物的性质受到配体和中心金属离子的影响。
配合物的颜色、溶解度、稳定性以及一些化学反应都与配体和金属离子的性质密切相关。
例如,某些过渡金属离子与氮、氧等电负性较高的配体形成的配合物具有较强的酸性;而某些具有大的络合度的配合物则具有较好的溶解性和稳定性。
五、应用无机配位化合物在化学、材料和生物领域具有广泛的应用。
在催化剂中,配合物的金属离子可以提供活性位点,从而促进化学反应的进行。
在生物医学中,金属配合物可以用作药物,通过与特定的生物分子相互作用来治疗疾病。
此外,配位化合物也广泛应用于材料科学领域,用于制备光电材料、磁性材料、液晶材料等。
六、进展与展望近年来,随着科学技术的不断发展,无机化学中的配位化合物在结构设计、属性调控以及应用领域方面取得了许多重要的进展。







第十八章配位化合物的价键理论§本章摘要§1.配位化合物的基本概念配位化合物配合物的命名异构现象2.配位化合物的稳定性酸碱的软硬分类影响配位单元稳定的因素3.配合平衡配合-解离配合配合平衡的移动4.配位化合物的价键理论配合物的构型与中心的杂化方式中心杂化轨道的形成价键理论中的能量问题价键理论的实验根据5.配合物的晶体场理论晶体场中的 d 轨道过渡金属化合物的颜色晶体场稳定化能(CFSE) Jahn - Teller 效应§1.配位化合物的基本概念一.配位化合物1 定义由中心原子 ( 或离子 ) 和几个配体分子(或离子)以配位键相结合而形成的复杂分子或离子,通常称为配位单元。
凡是含有配位单元的化合物都称做配位化合物,简称配合物,也叫络合物。
, Ni(CO)4都是配位单元,分别称作配阳离子、配阴离子、配分子。
[ Co(NH3)6]Cl3,K 3[Cr(CN)6], Ni(CO)4都是配位化合物。
[Co(NH3)6 ][Cr(CN)6] 也是配位化合物。
判断的关键在于是否含有配位单元。
2 构成内界是配位单元,外界是简单离子。
又如 K3[Cr(CN)6] 之中,内4 多基配体和螯合物乙二胺 H2N- CH2- CH2- NH2(表示为en ),其中两个氮原子经常和同一个中心配位。
象这种有两个配位原子的配体通常称双基配体 ( 或双齿配体) 。
而乙二胺四乙酸 ( EDTA ) ,其中 2 个N ,4 个 -OH 中的 O 均可配位,称多基配体。
负离子多基配体和正离子中心形成的中性配位单元,称为内盐。
和可形成内盐:界,外界是K+。
可以无外界,如Ni(CO)4。
但不能没有内界。
内外界之间是完全电离的。
内界配位单元又由中心和配体构成。
中心:又称为配合物的形成体,多为金属离子,尤其是过渡金属离子。
配体:经常是阴离子或分子。
3 配位原子和配位数配体中给出孤对电子与中心直接形成配位键的原子,叫配位原子。
第十八章配位化合物的价键理论§本章摘要§1.配位化合物的基本概念配位化合物配合物的命名异构现象2.配位化合物的稳定性酸碱的软硬分类影响配位单元稳定的因素3.配合平衡配合-解离配合配合平衡的移动4.配位化合物的价键理论配合物的构型与中心的杂化方式中心杂化轨道的形成价键理论中的能量问题价键理论的实验根据5.配合物的晶体场理论晶体场中的d 轨道过渡金属化合物的颜色晶体场稳定化能(CFSE) Jahn - Teller 效应§1.配位化合物的基本概念一.配位化合物,配位单元,称为内盐。
和界,外界是内界配,中心个N 原子与配位。
二配合物的命名文字母表次序,如和,则在前。
三异构现象1°电离异构内外界之间是完全电离的。
内外界之间交换成份得到的配合物,与原来的配合物互为电离异构。
它们电离出的离子种类不同,如[ CoBr(NH3)5]SO4和[CoSO4(NH3)5]Br ,前者可以使沉淀,后者则使Ag+沉淀。
H2O 经常做为配体,也经常在外界。
由于H2O 分子在内外界不同造成的电离异构,称为水合异构。
如[Cr(H2O)6]Cl3和[CrCl(H2O)5]Cl2·H2O 。
2°配位异构内界之间交换配体,得配位异构。
如[Co(NH3)6][Cr(CN)6] 和[Cr(NH3)6] [Co(CN)6]3°键合异构组成相同,但配位原子不同的配体,称两可配体,如-NO2-和-ONO-。
[Co(NO2)(NH3)5]Cl2和[ Co(ONO)(NH3)5]Cl2则互为键合异构。
总之,配体数目越多,种类越多,异构现象则越复杂。
2°旋光异构配体的相互位置关系不一致形成几何异构,当相互位置的关系亦一致时,也可能不重合。
比如人的两只手,互为镜像,各手指、手掌、手背的相互位置关系也一致,但不能重合。
互为镜像的两个几何体可能重合,但只要能重合则是一种。
若两者互为镜像但又不能重合,则互为旋光异构。
旋光异构体的熔点相同,但光学性质不同。
互为旋光异构体的两种物质,使偏2 空间异构(立体异构)键联关系相同,但配体相互位置不同,是空间异构的特点。
1°几何异构(顺反异构)配位数为4 的平面正方形结构顺式称顺铂,是抗癌药物,反式则无药效。
正方形的配合物M2a 2b,有顺反异构,M a 3b,不会有顺反异构。
正四面体结构,不会有顺反异构。
配为数为6 的正八面体结构振光偏转的方向不同。
按一系列的规定,定义为左旋、右旋。
不同的旋光异构体在生物体内的作用不同。
§2.配位化合物的稳定性一.酸碱的软硬分类软酸: 体积大,半径大,交界酸:等,等等交界碱:等二影响配位单元稳定的因素1 中心与配体的关系2 螯合效应螯合物稳定,以5 元环、6 元环螯合物最为稳定。
一般不形成配合物,但可以与乙二胺四乙酸(EDTA)形成螯合物。
六配位,2个N,4个O为配位原子,形成五个5元环,正八面体。
螯合物获得特殊稳定性的热力学原因:[L-M-L] + L-L = [M=L2] + 2L从简单配合物向螯合物转变过程中:由反应前的两个分子生成产物的3个分子.即这种螯合物的成环作用, 使体系的反应熵加大了, 正是这种成环后体系分子数增多而引起的熵增加, 增强了螯合物的稳定性.§3.配合平衡一.配合-解离平衡这个常数的值越大,表示配合反应进行得越彻底,配合物越稳定,故称之为K稳稳定得多。
在络合平衡的体系中,哪种络离子多?设平衡时[NH3]=1 . 根据各步的平衡方程式, 由K不稳越大,离解反应越彻底,配离子越不稳定,配位单元的形成可以认为是分步进行的,如配位单元的形成可以认为是分步进行的,如:K1、K2、K3、K4称为逐级稳定常数。
反应(1) 最易进行,反应(2)中的NH3受到第一个NH3的斥力,同时也有空间位阻,故难些。
(3)、(4)更难些。
这可从K1 > K2 > K3 > K4看出。
Kn 逐级减小,尤其是带电荷的配体。
所以,是体系中占主导多数的离子。
例1将0.2 的AgNO3溶液和2的NH3·H2O 等体积混合,求平衡后体系中的[ Ag+ ] 。
解: 由于体积变化,Ag+的起始浓度是0.1,而NH3的起始浓度是1。
而消耗NH3的浓度为0.1 x 2 = 0.2,故平衡时[ NH3 ] = ( 1 - 0.2 ),而设[ Ag+ ] = x, 则有:二配合平衡的移动1 与酸碱电离平衡的关系以(a) 为例讨论。
若pH 值增大,一方面由于[OH-] 增大,使反应进行,从而[ Ag+ ] 减小,导致(a) 左移;另一方面由于[ OH- ] 增大,反应逆向进行,从而[NH3] 增大,使(a) 右移。
可见,pH 的影响是很复杂的,因此酸碱电离平衡对配合-离解平衡的影响在分析化学中讲授。
2. 和沉淀-溶解平衡的关系络合剂、沉淀剂都可以和结合,生成配合物、沉淀物,故两种平3. 和氧化-还原反应的关系这种关系体现在半反应的值和值上。
氧化型+z e ——还原型,若氧化型被络合,值减小;还原型被络合,值增大。
若氧化型和还原型同时被络合,则计算更复杂些。
例3 = 0.34V的解法1: 将/ Cu 的看作Cu2+ / Cu 的非标准值,即[NH3 ] = [] = 1时的值.衡的关系实质是络合剂与沉淀剂争夺的问题,当然要和Ksp、K稳的值有关。
例2计算AgCl 在NH3·H2O 中( 6 ) 的溶解度,已知解:反应为:总反应为:Ksp 很小,K稳很大,Ag+不是结合成AgCl, 就是络合成,故游离的Ag+ 浓度极小. 可以认为从AgCl 中溶解下来的Ag+ , 完全变成,故[Cl-] = ,设其为x ,则消耗掉[NH3 ] 为2 x ,则[NH3]平= 6 - 2x 解法2: 将各反应视为独立反应, 用热力学函数(Gibss函数)和K稳以及半反应的相联系, 可达到同样的计算目的.等式两侧同时除以-2F, 得:AgCl 的,计算表明, AgCl可溶于6的NH3·H2O 中。
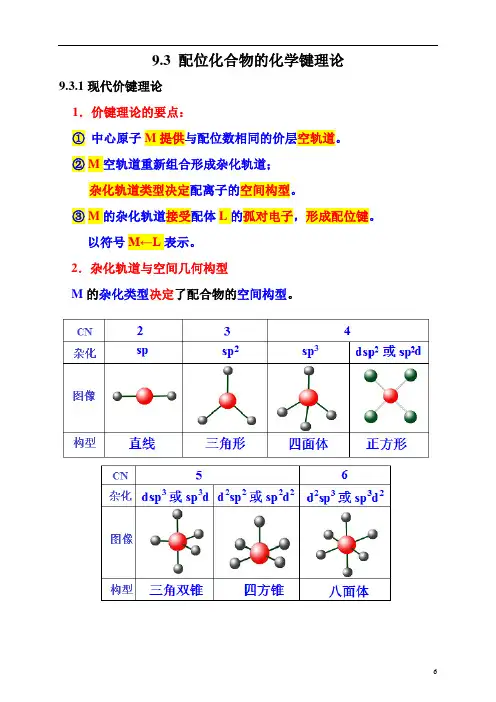
但AgI 的,在6 的NH3·H2O 中,其溶解度为,即AgI 不溶于NH3·H2O.§4.配位化合物的价键理论一.配合物的构型与中心的杂化方式二中心杂化轨道的形成1 个4s 空轨道,3 个4p 空轨道和2 个4d 空轨道形成sp3d2杂化轨道,正八面体分布。
6 个F-的6 对孤对电子配入sp3d2空轨道中,形成正八面体构型的配合单元。
例2 Ni(CO)4的成键情况在配体CO 的作用下,Ni 的价层电子重排成3d104s0形成sp3杂化轨道,正四面体分布,4 个CO 配体与sp3杂化轨道成配键,形成的Ni(CO)4构型为正四面体。
例3 讨论的成键情况形成d2sp3杂化,使用2 个3d 轨道, 1 个4s 轨道,3个4p 轨道。
用的内层d 轨道。
形成的配离子为正八面体构型。
空出1 个内层d 轨道,形成dsp2杂化轨道,呈正方形分布。
故构型为正方形。
例3 和例4 中,杂化轨道均用到三价键理论中的能量问题成一个对,能量升高一个p,p 叫成对能。
如中的d 电子,由变成,成2 个电子四价键理论的实验根据上可以测出物质的磁矩, 和单电子数n 有如下关系:式中B.M.是的单位,称为波尔磁子。
若测得= 5 B.M. , 可以推出n = 4 。
测出磁矩,推算出个中等强度的配体,在中究竟发生重排还是不发生重实验测得= 0 B.M. 推出n = 0,无单电子。
故NH3在此是强配体。
杂化轨道是d2sp3,正八面体,内轨配合物.测得的=5.88 B.M. ,推出n = 5 ,F-不使的d 电子重排。
所以磁§5.配合物的晶体场理论一.晶体场中的d 轨道1. 五种简并的d 轨道五种3 d 轨道,n = 3,l = 2,只是磁量子数目m 不同,在自由原子中能量简并。
当原子处于电场中时,受到电场的作用,轨道的能量要升高。
若电场是球形对称的,各轨道能量升高的幅度一致。
如图所示:3 °正方形场坐标原点位于正方形中心,坐标轴沿正方形对角线伸展。
显然分裂能q 。
若处于非球形电场中,则根据电场的对称性不同,各轨道能量升高的幅度可能不同,即原来的简并轨道将发生能量分裂。
2 晶体场中的d 轨道*六配位的配合物中,六个配体形成正八面体的对称性电场;*四配位时有正四面体电场、正方形电场。
尽管这些几何图形对称性很高,但均不如球形电场的对称性高。
中心的d 轨道在这些电场中不再简并。
1 °八面体场六个配体沿x、y、z 三轴的正负6 个方向分布,以形成电场。
在电场中各轨道的能量均有所升高。
但受电场作用不同,能量升高程度不同。
3 影响分裂能大小的因素1 °晶体场的对称性前面提及△p > △O > △T2 °中心离子的电荷数中心离子电荷数大,中心与配体距离近,则作用强,△大3 °中心原子所在的周期数第四周期过渡元素的△小,五、六周期的△相对大些。
4 °配体的影响( △递增次序)一般规律是:配位原子卤素< 氧< 氮< 碳,这个顺序称之为光化学序列,因为它影响△,而△的大小直接的波瓣与六个配体正相对,受电场作用大,能量升高得多,高于球形场。
;不与配体相对,能量升高的少,低于球形场。
高能量的、统称轨道;能量低的统称轨道,和能量差为,称为分裂能,八面体场中称为△O。
2 °正四面体场坐标原点为正四面体的中心,三轴沿三边方向伸展,4 个配体的位置如影响配合物的光谱。
4 分裂后的d 轨道中电子的排布某过渡金属d4组态,在八面体场中,d 电子的排布如何?究竟如何排列,取决于p 和△的大小关系:若△> p ,取甲种方式;若△< p 取乙种方式。
甲种:自旋成单电子的数目较低,称为低自旋方式。
乙种:自旋成单电子的数目较高,称为高自旋方式。
从光化学序列中看出等△大,常导致△> p ,取低自旋方式;而X-、OH-、H2O 等则△< p,常取高自旋方式。
具体配合物中,△和p 的大小是有能量数据的。
图所示,形成电场。
在正四面体场中,,受电场作用小,能量低于球形场;而受电场作用较大,能量高于球形场。
但显然两组轨道的差别较小。
于是其分裂能△T比八面体场的△O小得多。
在晶体场理论中,△和p 的值常用波数的形式给出。
波数是1cm 的长度相当于多少个波长。
可见波数越大,波长越小,频率越高,据E = hv,则能量越高。
总之,波数大,则能量高。
例如:中△= 10400,p = 15000,△< p组态,高自旋:表示为。
又如Fe(CN)64-,△= 33800 cm-1,p = 15000 cm-1,△> p 低自旋。