YYT0287-2017医疗器械生产企业内审检查表-销售部
- 格式:doc
- 大小:56.50 KB
- 文档页数:4

产品防护控制程序
1. 目的
对产品的标识、搬运、包装、贮存和保护过程进行控制,以维护产品固有质量,确保用户得到合格产品。
2. 适用范围
适用于原材料、过程产品和成品的标识、搬运、包装、贮存和保护。
3. 职责权限
3.1 技术部负责包装设计,负责提出防护性标识要求。
3.2 采购部负责原材料库和成品库的产品防护,负责向顾客交付产品过程中对产品进行防护。
3.3 生产部负责生产过程中各类产品的标识、搬运、包装、贮存和保护等过程的防护。
4. 工作程序
4.1 标识
4.1.1 产品包装或存储包装、货架上应有产品信息标识,包括名称、规格、数量、生产日期和存储条件或有效期等信息,以便正确、迅速识别和区分产品。
4.1.2 产品在中转、暂存和生产过程中应有状态标识,标明产品处于“待检”、“待判”、“合格”和“不合格”等状态。
4.2搬运
为防止原材料、过程产品及成品受到损坏影响质量,应依照以下原则进行作业。
4.2.1 车间物料、工序产品、成品搬运过程中,如应注意保护好产品及其标识,防止产品受到损害或标识遗失、损坏、入仓后应分类归位摆放。
4.2.2 内部搬运应选择合适的工位器具以确保产品不受外伤和污染,应采取轻拿轻放的原则,并且应符合物料进出洁净车间的相关要求。
搬运期间应注意周围事物,防止产品受到碰撞、摩擦,搬运期间的人员应经过适当的培训。
4.2.3 如因搬运不当造成产品损坏时,应立即报告责任部门负责人进行处理,并分析原因,采取纠正和预防措施。
质量部负责对该批处理后的产品重新进行检验。

审核实施计划
编号:XXXX-XX-XX
1.审核组组长:A 组员:B
2.审核时间:XX年X月X日至XX年X月X日
3.审核目的:
(1)检查本公司的质量管理体系运行的符合性和有效性;
(2)检查新修订质量体系文件是否符合YY/T 0287-2017/ISO 13485:2016、《医疗器械生产质量管理规范》及现场检查指导原则的要求;
(3)完善公司质量管理体系。
4. 审核依据:
(1)YY/T 0287-2017/ISO 13485:2016《医疗器械质量管理体系用于法规的要求》(简称“标准”);
(2)《医疗器械生产质量管理规范》(国家食品药品监督管理局令第64号)、《医疗器械生产质量管理规范现场检查指导原则》(食药监械监〔2015〕218号附件1)等相关法律法规;(3)公司质量管理体系文件;
(4)产品技术文件;
(5)公司的有关合同。
5. 现场审核期间请被审核方有关人员参加下列活动:
首末次会议:管理者代表、内审员全体成员及各部门负责人参加;
审核活动:按审核日程安排,被审核方有关人员在本岗位。
编制:审批:日期:
内审检查表
不符合项报告
内部质量管理体系审核报告
编制:审批:日期:。
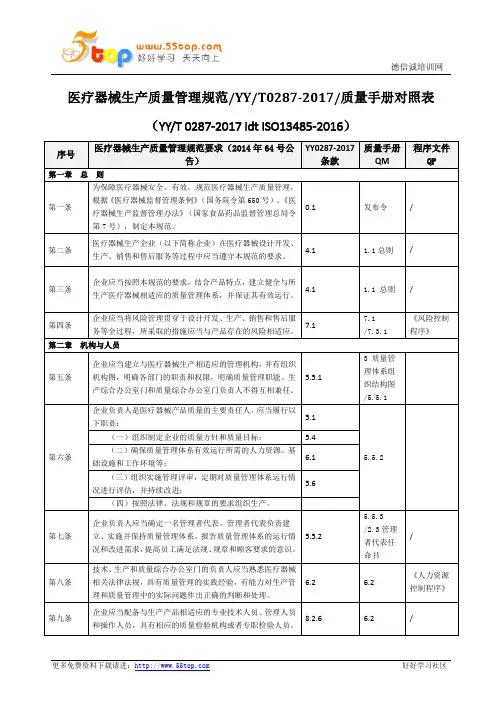
医疗器械生产质量管理规范/YY/T0287-2017/质量手册对照表(YY/T 0287-2017 idt ISO13485-2016)序号医疗器械生产质量管理规范要求(2014年64号公告)YY0287-2017条款质量手册QM程序文件QP第一章总则第一条为保障医疗器械安全、有效,规范医疗器械生产质量管理,根据《医疗器械监督管理条例》(国务院令第650号)、《医疗器械生产监督管理办法》(国家食品药品监督管理总局令第7号),制定本规范。
0.1 发布令/第二条医疗器械生产企业(以下简称企业)在医疗器械设计开发、生产、销售和售后服务等过程中应当遵守本规范的要求。
4.1 1.1总则/第三条企业应当按照本规范的要求,结合产品特点,建立健全与所生产医疗器械相适应的质量管理体系,并保证其有效运行。
4.1 1.1 总则/第四条企业应当将风险管理贯穿于设计开发、生产、销售和售后服务等全过程,所采取的措施应当与产品存在的风险相适应。
7.17.1/7.3.1《风险控制程序》第二章机构与人员第五条企业应当建立与医疗器械生产相适应的管理机构,并有组织机构图,明确各部门的职责和权限,明确质量管理职能。
生产综合办公室门和质量综合办公室门负责人不得互相兼任。
5.5.13 质量管理体系组织结构图/5.5.1第六条企业负责人是医疗器械产品质量的主要责任人,应当履行以下职责:5.15.5.2(一)组织制定企业的质量方针和质量目标; 5.4(二)确保质量管理体系有效运行所需的人力资源、基础设施和工作环境等;6.1(三)组织实施管理评审,定期对质量管理体系运行情况进行评估,并持续改进;5.6(四)按照法律、法规和规章的要求组织生产。
第七条企业负责人应当确定一名管理者代表。
管理者代表负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高员工满足法规、规章和顾客要求的意识。
5.5.25.5.3/2.3管理者代表任命书/第八条技术、生产和质量综合办公室门的负责人应当熟悉医疗器械相关法律法规,具有质量管理的实践经验,有能力对生产管理和质量管理中的实际问题作出正确的判断和处理。

文件编号:CMHZ-SC-01控制状态:受控 非受控□版本号:000发放编号:CMHZ-2017-001质量手册编制:审核:批准:2017年9月29日发布 2017年9月29日实施XXX公司目录1.范围 (6)总则 (6)删减和不适用说明 (6)不适用条款说明: (6)医疗器械生产质量管理规范不适用条款说明: (6)引用的法规和标准 (7)质量手册的管理 (8)质量方针与质量目标 (9)质量方针: (9)质量目标: (9)2 企业概况 (11)修改页 (12)颁布令 (13)管理者代表任命书 (14)公司管理架构图 (14)3 质量管理体系组织结构图 (15)4.质量管理体系 (16)总要求 (16)文件要求 (18)总则 (18)质量手册 (19)医疗器械文档 (19)文件控制 (20)记录控制 (20)支持性文件 (21)5.管理职责 (21)管理承诺 (21)以客户为关注焦点 (21)质量方针 (21)策划 (22)质量目标 (22)质量管理体系策划 (22)职责、职权与沟通 (23)职责与权限 (23)总经理 (24)管理者代表 (25)综合办公室 (25)品保部: (26)技术部 (27)运营部 (28)市场部 (28)销售部 (29)内部沟通 (29)管理评审 (30)总则 (30)评审输入 (30)评审输出 (30)支持性文件 (31)6.资源管理 (32)提供资源 (32)人力资源 (32)基础设施 (32)工作环境和污染的控制 (33)工作环境 (33)支持文件 (33)7.产品实现 (34)产品实现的策划 (34)适用范围 (34)策划内容 (34)质量计划 (34)与顾客有关的过程 (35)产品要求的确定 (35)产品要求的评审 (35)沟通 (36)设计和开发 (36)总则 (36)设计和开发策划 (36)设计和开发输入 (36)设计和开发输出 (37)设计和开发评审 (37)设计和开发验证 (37)设计和开发确认 (38)设计和开发的转换 (38)设计和开发更改的控制 (38)设计和开发文档 (38)风险管理 (38)采购 (39)采购过程 (39)采购信息 (39)采购产品的验证 (39)生产和服务提供 (40)生产和服务提供控制 (40)产品的清洁 (40)服务活动 (41)无菌医疗器械的专用要求 (41)生产和服务提供过程的确认 (41)灭菌过程和无菌屏障系统确认的专用要求 (41)标识 (41)可追溯性 (42)顾客财产 (43)产品防护 (43)监视和测量设备的控制 (43)相关文件 (44)8测量、分析和改进 (44)总则 (44)监视和测量 (44)反馈 (44)投诉处置 (45)向监管机构报告 (45)内部审核 (45)过程监视和测量 (46)产品监视和测量 (46)不合格产品控制 (46)总则 (46)交付前发现不合格品的相应措施 (47)交付后发现的不合格品的响应措施 (47)返工 (47)数据分析 (47)改进 (48)总则 (48)纠正措施 (48)预防措施 (48)附件一程序文件清单 (50)附件二职能分配表 (51)附件三医疗器械生产质量管理规范/YY/T0287-2017/质量手册对照表 (52)1.范围总则本手册是依据YY/T 0287-2017/ISO13485:2016《医疗器械质量管理体系用于法规的要求》和GB/T 19001-2016/ISO 9001:2015《质量管理体系要求》标准制定。
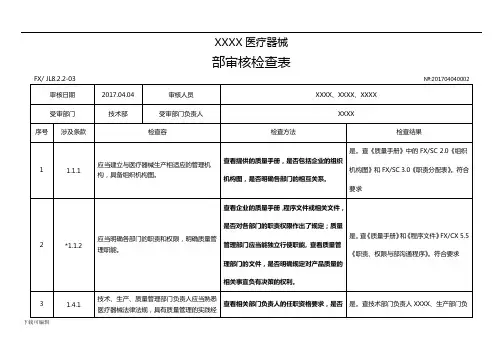
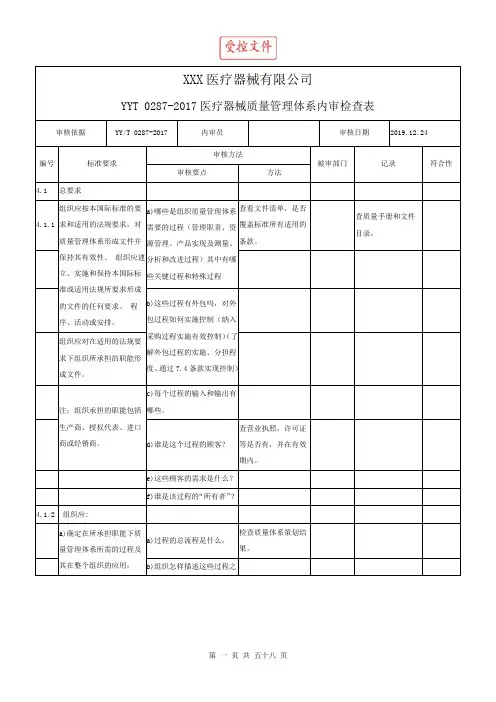

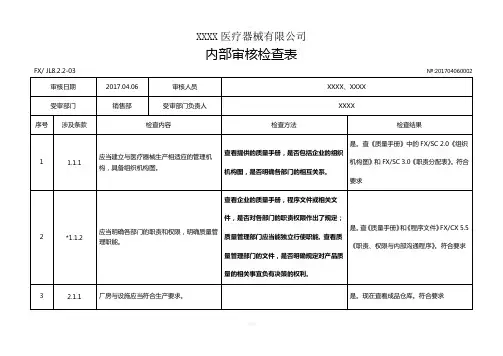
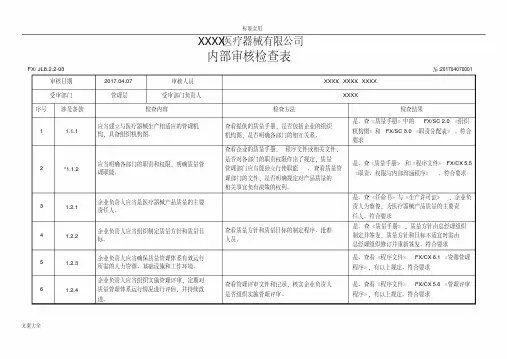
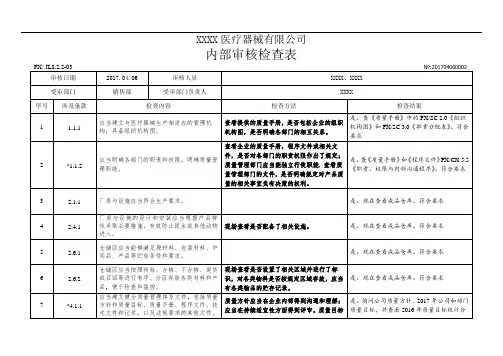
XXXX医疗器械有限公司内部审核检查表FX/ JL8.2.2-03审核日期2017.04.07审核人员受审部门管理层受审部门负责人№:201704070001 XXXX、 XXXX、 XXXXXXXX序号涉及条款检查内容1 1.1.1 应当建立与医疗器械生产相适应的管理机构,具备组织机构图。
2 *1.1.2 应当明确各部门的职责和权限,明确质量管理职能。
3 1.2.1 企业负责人应当是医疗器械产品质量的主要责任人。
4 1.2.2 企业负责人应当组织制定质量方针和质量目标。
5 1.2.3 企业负责人应当确保质量管理体系有效运行所需的人力资源、基础设施和工作环境。
企业负责人应当组织实施管理评审,定期对6 1.2.4 质量管理体系运行情况进行评估,并持续改进。
7 *1.2.5 企业负责人应当确保企业按照法律、法规和规章的要求组织生产。
检查方法查看提供的质量手册,是否包括企业的组织机构图,是否明确各部门的相互关系。
查看企业的质量手册,程序文件或相关文件,是否对各部门的职责权限作出了规定;质量管理部门应当能独立行使职能, 查看质量管理部门的文件,是否明确规定对产品质量的相关事宜负有决策的权利。
查看质量方针和质量目标的制定程序、批准人员。
查看管理评审文件和记录,核实企业负责人是否组织实施管理评审。
检查结果是。
查《质量手册》中的 FX/SC 2.0 《组织机构图》和 FX/SC 3.0 《职责分配表》。
符合要求是。
查《质量手册》和《程序文件》 FX/CX 5.5《职责、权限与内部沟通程序》。
符合要求是。
查《任命书》与《生产许可证》,企业负责人为蔡俊,为医疗器械产品质量的主要责任人。
符合要求是。
查《质量手册》,质量方针由总经理组织制定并签发,质量方针和目标不适宜时需由总经理组织修订并重新签发。
符合要求是。
查看《程序文件》 FX/CX 6.1 《资源管理程序》,有以上规定。
符合要求是。
查看《程序文件》 FX/CX 5.6 《管理评审程序》,有以上规定。
忠告性通知控制程序
1.目的
为确保产品在交付后,使顾客正确防护、贮存、使用产品,当发生任何形式的改动或质量问题时,能迅速采取纠正和预防措施,并及时以电话、电报、传真、信函或公告形式通知有关顾客,并在必要时实行产品追回。
2.范围
适用于公司所有产品忠告性事件的信息发布、反馈和处理。
3.职责
3.1质量部负责产品忠告性通知的拟定,总经理批准;
3.2销售部负责忠告性通知发布,产品的召回;
3.3管理者代表对符合不良事件报告准则的忠告性通知事项向主管机构报
告;
3.4质量部、技术部对召回产品实施改善,生产部协助。
4.工作程序
4.1定义
4.1.1忠告性通知
在医疗器械交付后,由组织发布的通知,旨在以下方面给出补充信息和/或建议宜采取的措施:(注:忠告性通知的发布要符合国家或地区法规) 医疗器械在使用时应注意的补充事宜;
医疗器械的改动,如结构、电路的改动、电源或环境的改动;
医疗器械返回组织或经销商;
医疗器械的销毁。
4.2产品交付后发现未达到预期用途及可能对病人或操作人员的伤害或潜
在的伤害或不符合法规要求时,由质量部组织,总经理、销售部、技术和生产部参与进行质量分析会,研讨是否发布忠告性通知,包括实行产品召回并报告当地或国家行政主管部门。
4.3忠告性通知或产品召回应包括如下内容:。
设计开发控制程序
1. 目的
对产品设计和开发过程实施策划和控制,确保公司设计开发过程管理有序,项目实施准确有效,产品满足顾客和法规的要求。
2. 适用范围
适用于公司产品设计开发的全过程,设计和开发过程分为:策划、输入、输出、评审、验证、确认、设计转换活动、更改过程以及设计开发文档等。
3. 职责权限
3.1总经理负责组织建立产品设计和开发项目组,指定项目负责人,并提供开发新产品所需的人力资源,基础设施和工作环境。
项目负责人负责设计开发策划。
3.2销售部负责市场调研,提供市场需求信息;评估或提出产品及产品的功能、性能、外观及其他要求;针对新项目评估或提出产品销售量、销售价格等销售信息。
3.2技术部负责设计和开发输入、输出、评审、验证和确认工作,对设计开发全过程组织和实施。
3.3采购部负责新产品物料的采购;供应商的选择和审核。
3.4生产部负责设计开发转换活动,样品的实现和工艺验证。
3.5质量部负责新产品的检验和试验,参与设计评审、验证、确认、风险管理等控制活动。
4. 工作程序
4.1设计和开发的策划
新产品立项后,由项目负责人负责编制《设计开发策划书》,报总经理批准。
策划内容包括:
a)设计和开发项目的目标和意义的描述,技术指标分析;
b)确定设计和开发各阶段,以及适合于每个设计和开发阶段的评审、验证、确认和设计转换活动;
c)识别和确定各个部门设计和开发的活动和接口,明确各阶段的人员或组织的职责、评审人员的组成,以及各阶段预期的输出结果;。
YY/T 0287-2017 标准与我国法规(医疗器械)对
8.3.2 交付前发现不合格品的响应措施《生产规范》64号公告第六十八条
10.1.1
*10.2.1
8.3.3 交付后发现不合格品的响应措施《生产规范》64号公告第六十九条
10.3.1
*11.5.1 对已交付的不合格品的响应措施及
向监管机构报告的具体要求
8.3.4 返工《生产规范》64号公告第七十条10.4.1 10.4.2
8.4 数据分析《生产规范》64号公告第七十三条11.3.1 明确收集的质量数据还包括不良事件信息
8.5 改进
8.5.1 总则《生产规范》第七十四条
8.5.2 纠正措施《生产规范》第七十四条11.4.1 8.5.3 预防措施《生产规范》第七十四条11.4.2
械)对应关系。