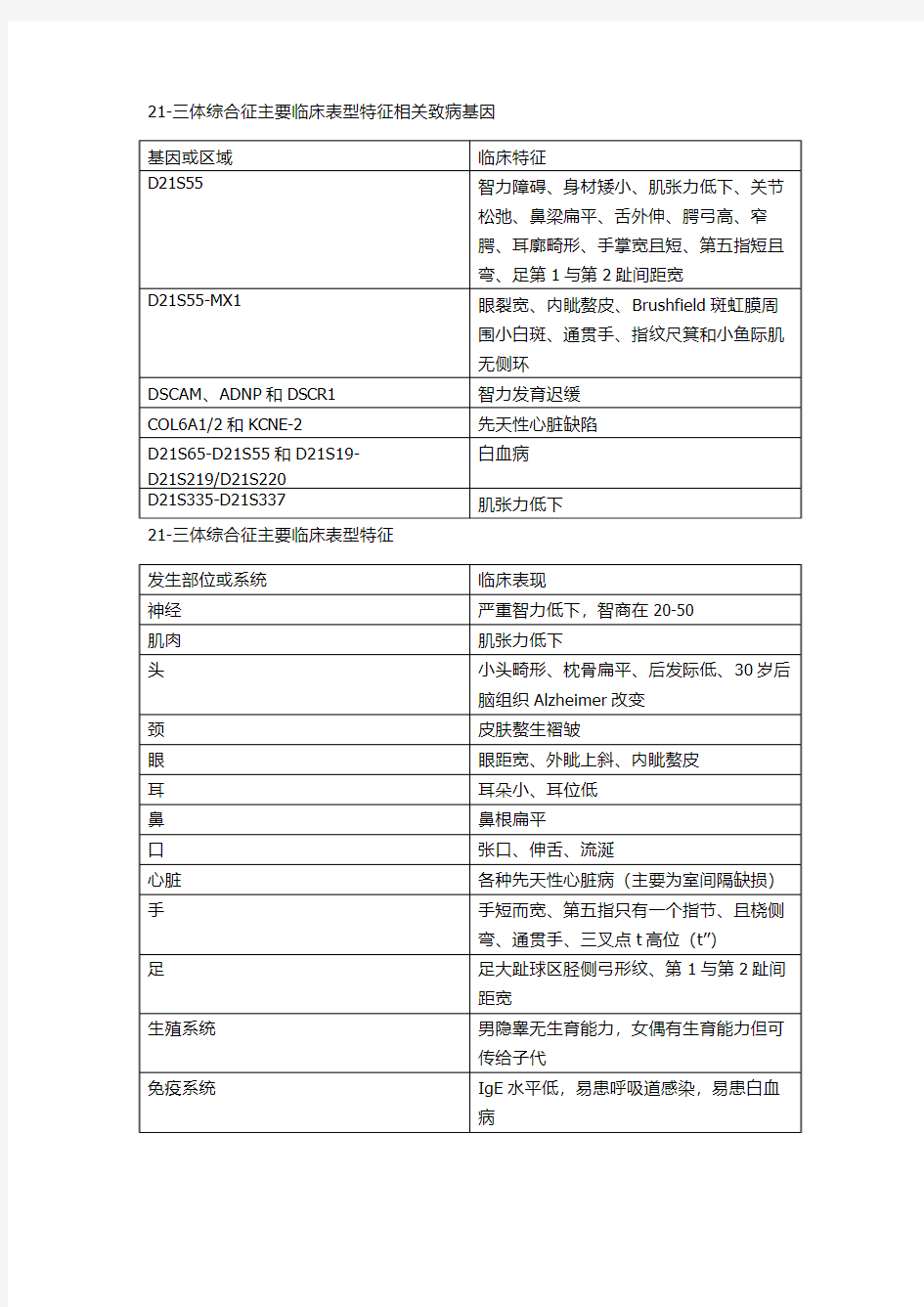

21-三体综合征主要临床表型特征相关致病基因
21-三体综合征主要临床表型特征
Angelman综合征相关致病基因
1、15q11-13区域缺失;
2、15号染色体的父源同源二倍体;
3、印迹基因功能缺失。AS是由母源染色体15q11-13上编码泛素蛋白连接酶E3的UBE3A基因缺失或表达异常所致。UBE3A基因在印记基因调控下在人体不同组织表达具有差异性[9],正常颅脑组织内母源UBE3A基因表达活跃,父源UBE3A基因不表达[14]。由于AS患者母源染色体15q11~13上的UBE3A基因缺失或表达异常,颅内无编码泛素蛋白连接酶E3的UBE3A基因表达,因此AS患者黑质、纹状体、海马及小脑浦肯野细胞蛋白泛素化异常[15]。然而泛素化异常如何导致AS患者一系列临床表现尚无定论。Mulherkar等[16]认为,泛素蛋白连接酶E3介导了多巴胺神经元突触前、后膜酪氨酸羟化酶的降解,AS患者因为缺乏该酶,致使黑质纹状体通路功能障碍,从而出现运动障碍、共济失调。这一理论从神经生物学角度揭示了泛素化异常致运动障碍的机制。近来Proville等[17]提出小脑-皮层网络假说,认为小脑能发放电冲动调节大脑皮层电活动。有研究者在AS小鼠泛素化异常的小脑浦肯野细胞检测到自发性160 Hz电活动,这种电活动可改变大脑皮层的长时程抑制效应,从而破坏大脑皮层兴奋与抑制平衡。而AS患者的智力低下、语言障碍、癫痫发作等核心症状均为大脑功能异常的表现。因此推测,异常泛素化的浦肯野细胞通过小脑-皮层网络改变了大脑电活动,导致AS患者出现一系列临床表现[18,19]。该假说从电生理学角度揭示了小脑泛素化异常致智力低下、语言障碍、癫痫发作的机制。以上研究表明,UBE3A基因缺陷所致泛素化异常可通过多种机制导致AS患者出现运动障碍、共济失调、语言障碍、癫痫发作等临床表现。
基因缺陷分型
UBE3A基因的缺失或表达异常由4种不同方式导致:母源15q11~13缺失、父源单亲二倍体(uniparental disomy,UPD)、印(imprinting centre,IC)缺陷及突变[9]。近年研究发现,不同基因缺陷型患者的临床表现具有较大差异。为了推动单基因缺陷病研究的发展,大量研究者对不同基因型患者的形成原因和临床表现进行了深入的研究[11,12]。
Angelman综合征主要临床表现
共同特征(100%) 严重发育迟缓
平衡障碍:常表现为共济失调以及四肢震颤
运动障碍:步态不稳,程度不重
行为特征:频繁大笑、明显的兴奋动作、易激惹、常伴拍手或舞动动作、多动
语言障碍:无或极少量词汇,非语言交往能力强于语言能力
常见表现(80%)
头围增长落后,随访至2岁仍表现为小头畸形,多见于缺失型患者癫痫发作,常3岁前起病,病情可随年龄增长缓解但持续存在于整个童年时期特征性脑电图:可先于癫痫发作出现
相关症状(20%~80%)
枕部扁平;喜吐舌;吸吮或吞咽障碍;婴儿期喂养困难;巨大下颌;牙间隙宽;频繁流涎;过度咀嚼动作;斜视;皮肤色素减退,与家人相比头发和眼睛颜色浅(仅见于缺失型);下肢过度活动,腱反射亢进;上举或弯曲上肢,尤其是行走时;热敏感增强;异常睡眠觉醒周期;迷恋水;肥胖;脊柱侧凸;便秘
新生儿、婴儿期临床特征:出生时通常正常,很快出现严重的发育迟缓。幼年早期患者发育迟缓,出现无意识发笑伴欢乐姿势、共济失调、震颤及小头畸形。
癫痫:可以始于1岁后,但通常都于3岁前出现,严重程度不一,癫痫发作可在青春期减少或消失。
运动病态:各种运动发育标准迟缓,震颤开始于出生后6个月,患者有特征性共济失调步态伴失调前倾、双脚分开待跑姿势,提臂屈肘。
头面部畸形:出生时面部无异常,部分患者幼时出现小头畸形,可能继发性脑组织发育不良,大多数有后枕平坦,头短畸形。下颌骨大小正常,但面中部发育不良,常见上唇单薄、眼深陷、张口流涎、牙缝疏。
其他:婴儿期患儿通常手持物品,口含杂物,表现为好动症,该症状可以随年龄增大而改善。50%-70%的患儿可见毛发、皮肤和眼部色素减退。
William’s综合征相关致病基因
7号染色体长臂近着丝粒片段7q11.23微缺失。
William’s综合征主要临床表现
以心血管病变(主动脉瓣狭窄、肺动脉狭窄)、婴儿期高钙血症为主。
智力:轻度到中度智力低下,智商介于40-100之间,平均60;读写能力差,对简单的算术也感到困难;声音嘶哑;听觉过敏;注意力集中时间虽短,但对音乐、唱歌、弹奏乐器有惊人的耐力。
特发性个性:对人友善,健谈多语,喜欢社交。
生长发育:轻度产前生长迟缓,产后发育障碍,身高低于正常人。
颅面部:小头,眉部潮红,眼裂小,内眦赘皮,斜视,虹膜蓝色,眼周围皮下组织丰满;低鼻梁,鼻孔朝上;人中长;唇厚张口;牙釉质发育不良。
心血管:主动脉瓣狭窄是本病的特征性改变。此外,还可以有周围肺动脉狭窄、肺动脉瓣狭窄、房间隔缺损、肾动脉狭窄伴高血压。
泌尿系统:两侧肾脏大小不对称,位置异常,肾钙质沉着;膀胱憩室,尿道狭窄,膀胱输尿管回流。
躯干与四肢:关节活动受限;脊柱前变、侧弯或后弯;指甲发育不良;趾外翻,走路笨拙等。
部分患儿会出现短暂性高血钙。
苯丙酮尿症(PKU)相关致病基因
常染色体隐性遗传病。经典PKU主要原因是因苯丙氨酸羟化酶(PAH)缺陷或缺乏,
而PAH缺陷或缺乏是由PAH基因突变引起,PAH基因位于12q22-24。非经典PKU是因四氢生物蝶呤(BH4)缺陷或缺乏,而BH4缺陷或缺乏是由于BH4合成缺陷或再生缺陷引起。
苯丙酮尿症主要临床表现.
1.生长发育迟缓
除躯体生长发育迟缓外,主要表现为智力发育迟缓。表现为智商低于同龄正常儿,生
后4~9个月即可出现。重型者智商低于50,语言发育障碍尤为明显,这些表现提示
大脑发育障碍。
2.神经精神表现
近半数合并癫痫发作,其中约1/3为婴儿痉挛症,多在生后18个月以前出现。约80%有脑电图异常,可表现为高峰节律紊乱、灶性棘波等,癫痫发作可随年龄增长而变换
发作形式,绝大多数患儿有抑郁、多动和孤独症倾向等精神行为异常。神经系统异常
体征不多见,可有脑萎缩而有小脑畸形,肌张力增高,步态异常,腱反射亢进,手部
细微震颤和肢体重复动作。
3.皮肤毛发表现
由于酪氨酸酶受抑制,使黑色素合成减少,故患儿毛发色淡而呈发黄、皮肤和虹膜色浅。皮肤常干燥,易合并有湿疹和皮肤划痕症。
4.其他
由于苯丙氨酸羟化酶缺乏,苯丙氨酸经旁路代谢后产生苯丙酮酸和苯乙酸,从汗液和
尿中大量排出而有霉臭味(或鼠尿味)。
经典PKU和非经典PKU生后数月内不易区分。不同的是非经典PKU即使很早开始饮
食治疗,神经系统症状2-3月后仍可出现,且进行性恶化。
脊髓性肌萎缩症(SMA)相关致病基因
属常染色体隐性遗传病。SMA致病基因被定位于5q11.2-13.3区域,确定4个相关基因:运动神经元生存基因(SMN)、神经元凋亡抑制蛋白基因(NAIP)、基本转录因
子IiP44亚单位基因(BTF2P44)和H4F5基因。其中SMN缺失引起,则会导致SMA
发生。作为SMN的毗邻基因,NAIP缺失只代表缺失程度更大,没有足够证据证实NAIP是SMA的主要致病基因。BTF2P44亦不是SMA的决定基因,而H4F5被认为是SMA表型修饰基因。
脊髓性肌萎缩症主要临床表现
SMA分为三种类型,大多数患者为SMA-Ⅰ型,其次为Ⅱ型,Ⅲ型发病率最低。
1.婴儿型脊髓性肌萎缩
也称为SMA-I型或Werdning-Hoffmann病。本型为3型中最为严重的,部分病例在宫内发病,胎动变弱,半数在出生时或出生后的最初几个月即可发病,且几乎均在5个
月内发病,能存活1年者罕见。这些患儿在胎儿期已有症状,胎动减少,出生后即有
明显四肢无力,喂养困难及呼吸困难。临床特征表现:
(1)对称性肌无力,首先双下肢受累,迅速进展,主动运动减少,近端肌肉受累最重,不能独坐,最终发展为手足尚有轻微活动。
(2)肌肉弛缓,张力极低。患儿卧位时两下肢呈蛙腿体位、髋外展、膝屈曲的特殊体位,腱反射减弱或消失。
(3)肌肉萎缩,可累及四肢、颈、躯干及胸部肌肉,由于婴儿皮下脂肪多,故肌萎缩不易被发现。
(4)肋间肌麻痹,轻症者可有明显的代偿性腹式呼吸,重症者除有严重呼吸困难外,吸气时可见胸骨上凹陷,即胸式矛盾呼吸,膈肌运动始终正常。
(5)运动脑神经受损,以舌下神经受累最常见,表现舌肌萎缩及震颤。
(6)预后不良,平均寿命为18个月,多在2岁以内死亡。
2.中间型脊髓性肌萎缩
也称为SMA-Ⅱ型、中间型SMA或慢性SMA,发病较Ⅰ型稍迟,多于1岁内起病,进
展缓慢。患儿在6~8个月时生长发育正常,多数病例表现以近端为主的严重肌无力,下肢重于上肢。许多Ⅱ型患儿可独坐,少数甚至可以在别人的帮助下站立或行走,但
不能独自行走。多发性微小肌阵挛是主要表现。呼吸肌、吞咽肌不受累,面肌不受累,括约肌功能正常。本型具有相对良性的病程,生存期超过4年,可存活至青春期以后。
3.少年型脊髓性肌萎缩
又称SMA-Ⅲ型,也称为Kugelberg-Welander病、Wohlfart-Kugelberg-Welander综合征或轻度SMA,是SMA中表现最轻的一类。本病在儿童晚期或青春期出现症状,起病隐袭,进展缓慢,表现为神经元性近端肌萎缩。开始为步态异常,下肢近端肌肉无力,蹒跚步态,登楼和蹲下站起困难,渐累及下肢远端和双上肢,举臂困难。常伴腰椎前突,腹部凸起,腱反射可有可无。维持独立行走的时间与肌无力的发病年龄密切相关,2岁前发病者将在15岁左右不能行走,2岁后发病者可一直保持行走能力至50岁左右。可存活至成人期。
染色体异常基因检测 染色体 染色体是组成细胞核的基本物质,是基因的载体。人类体细胞有23对染色体,其中22对为男女所共有,称为常染色体;另外一对为决定性别的染色体,男女不同,称为性染色体;男性为XY,女性为XX。 染色体异常 体细胞或性细胞内染色体发生异常改变称为染色体异常,可分为数目异常和结构异常两大类。染色体异常可以自发地产生,称为自发突变;也可以通过物理的、化学的和生物的诱变作用而产生;还可以由亲代遗传所致。 染色体异常是导致出生缺陷、先天性遗传病、自然流产、不孕不育等的重要遗传因素。新生儿染色体异常的发病率为1/60。 常见的染色体微缺失/微重复综合征有300种,多会导致不同程度的发育异常和智力障碍,常伴有五官、内脏、四肢等方面的畸形,严重危害患者健康,给家庭和社会带来极大负担。 染色体异常基因检测 核子基因基于新一代高通量测序仪开发的染色体异常检测方法,可对流产组织、脐带血、绒毛、羊水、外周血等多种来源的样本进行测序分析,一次性检测23对染色体非整倍体、300种常见微缺失/微重复综合征及其他100kb以上的染色体异常,全面排查流产、胎儿异常、新生儿/儿童表型异常、不孕不育原因。 临床应用: 1、流产组织染色体异常检测:查明反复流产、异常妊娠的原因,指导再次生育。 2、胎儿染色体异常检测:排查胎儿异常的原因。 3、新生儿/儿童染色体异常检测:为发育异常、智力障碍,多发畸形等患儿排查染色体异常因素,为疾病确诊和治疗提供指导信息。 4、成人染色体异常检测:排查不孕不育、不良孕产史的遗传因素。 ????
技术优势: 1、全面覆盖:一次性检出23对染色体非整倍体和300种常见染色体微缺失/微重复综合征。 2、分辨率高:可检出100kb以上的染色体微缺失/微重复和低至5%的嵌合体。 3、准确度高:检测准确率>99% 4、自动解读:对检测到的微缺失/微重复,自动关联国际主流染色体异常数据库(Decipher、ISCA、OMIM、Clinvar)进行解读。 检测适用人群: 1、反复性流产,需要查明流产原因并确认再次生育方案的家庭。 2、超声显示结构异常或宫内发育迟缓的胎儿及父母。 3、临床表型为发育异常、智力障碍、多发畸形等患儿及父母。 4、具有染色体疾病家族史的夫妇及胎儿。 5、曾生育染色体疾病患儿的夫妇。 6、不孕不育或有不良孕产史的夫妇。 ????
基因诊断在单基因遗传病中的应用 【摘要】基因诊断是利用分子遗传学技术在DNA或RNA水平上对某一基因进行突变分析,从而对特定疾病进行诊断。基因诊断因其直接诊断性、高特异性、灵敏性、早期诊断性弥补了表型诊断的不足而被广泛应用。本文主要从基因诊断方法如核酸分子杂交、聚合酶链反应及相关技术、DNA序列测定、DNA芯片、连锁分析等在单基因遗传病中的应用进行综述。 【关键词】基因诊断;单基因遗传病;分子诊断;血友病 1基因诊断 基因诊断(gene diagnosis)又称DNA诊断或分子诊断,通过从体内提取样本用基因检测方法直接检测基因结构及其表达水平的改变,检测病原体基因型,进而判断是否有基因异常或携带病原微生物,或利用分子生物学技术从DNA水平检测人类遗传性疾病的基因缺陷。应用基因诊断技术可以针对已确诊或拟诊遗传性疾病的患者及其家系成员,根据遗传学的基本原理,通过分子生物学的实验手段检查被检个体相关基因的异常,确定隐形携带者状态及在症状出现前的疾病易感性等,从而达到临床确诊的目的。因此,基因诊断迅速在临床诊断领域特别在遗传病研究领域得到了较为广泛的应用。目前的基因诊断方法主要有核酸分子杂交、聚合酶链反应及相关技术、DNA序列测定、DNA芯片、连锁分析等。 2单基因遗传病 单基因遗传病是指由单个基因异常导致且以孟德尔方式遗传的疾病,是我国常见出生缺陷的重要原因之一,较为常见且研究较多的有血友病、苯丙酮尿症(PKU)、肝豆状核变性、地中海贫血等等。除部分单基因遗传病可通过手术加以矫正外,绝大部分遗传病是致死、致残、致畸性疾病,且目前均无法治疗,进行遗传性疾病的产前诊断,是避免致死、致残、致畸性疾病胎儿出生的重要手段。 3基因诊断的应用 3.1在B型血友病中的应用 血友病B(hemophilia B)是因凝血因子Ⅸ(FlX)基因缺陷引起的x-连锁隐性遗传出血性疾病,在男性中的发病率约为1/30000,散发率可达患者总数的30%-50%[1]由于目前还不能根治,对于携带者和高危胎儿进行基因诊断非常必要。血友病B基因缺陷类型十分繁多,基因缺陷包括缺失、插入和点突变,其中80%左右为单个碱基突变[2]。目前已发现的突变位点中,除了导致氨基酸序列改变的突变外,还发现不少的CpG区、剪切位点的突变[3]。常用于血友病B连锁分析的方法有限制性片段多态性(restriction fragment length polymorphisms,RFLP)
基因诊断在遗传病监测中的应用 目前发现人类遗传性疾病有3 000多种,如果仅依靠以往的染色体分析技术或对基因产物与代谢物的测定,我们只能对其中为数极少的一部分疾病在发病前或产前进行诊断。因为许多基因的表达有时相性和组织特异性(如有些基因在胎儿早期并不表达、苯丙氨酸羟化酶只在肝组织中表达)。用常规的方法采集的胎儿标本或其他人体材料,常常不能测出这些基因的产物或代谢产物。 然而,作为构成机体基本单位的细胞,无论其来自何种器官或组织,它们的基因组成却是完全一致的;虽然在某些特异化的组织细胞中某些基因并不表达,但那些基因的突变却存在于一切细胞之中。如果采用基因分析的方法进行监测,在个体发育的任何阶段,以任何一种有核细胞为检材,基因的缺陷都能被监测出来。 这就是近十几年来飞速发展的重组DNA技术给遗传病的早期(症状前和出生前)诊断带来的福音。重组DNA 技术不仅极大地丰富了我们对人类遗传病分子病理学的知识,而且同时也提供了从DNA水平对遗传病进行基因诊断的手段。自从1978年发现第一个限制酶切位点多态性并应用于遗传病(镰形细胞贫血)的基因诊断以后,能够进行基因诊断的病种不断增加,方法和途径越来越多。 一、基因突变的类型 造成基因突变的原因很多,有自发的也有外界理化因素的影响。从DNA序列改变的角度来看,不外乎单核苷酸的取代和DNA片段的插入或缺失两大类型。所产生的后果取决于突变发生的位置和性质,只要影响了基因表达过程中的任何一个环节,都会导致遗传性疾病。归纳起来如表1所示 表1 基因突变及效应一览表 DNA序列的改变突变发生的部位mRNA水平的表现基因产物的改变举例 1.大片段缺失或插入 整个基因缺如缺如α地中海盆血 基因片段异常功能缺陷DMD、BMD 2.少数核苷酸的缺失或插入 外显子与内含子接界拼接异常缺如 3的整数倍外显子缩短或延长异常(氨基酸缺失或插入)Hb Leiden 非3的整数倍外显子缩短或延长异常(移码突变)β地中海盆血 3.单核苷酸取代 启动子减少减少β地中海盆血 剪接信号剪接异常缺如β地中海盆血 PolyA信号不稳定减少β地中海盆血 密码子中性突变正常 密码子错义突变氨基酸取代异常血红蛋白 密码子或内含子剪接异常缺如或移码突变β地中海盆血 密码子无义突变肽链提前终止β地中海盆血 终止密码肽链延长,量减少Hb Canstant spring 起始密码β地中海盆血缺如β地中海盆血 二、遗传病基因诊断的途径 在了解了基因突变的各种类型之后,对应用何种方法来诊断它们便很容易理解了。例如某种遗传病是由于基因缺失造成的,可通过监测受检者是否缺失该基因来直接判断其基因型。如果某遗传病是核苷酸取代造成的点突变,便可以通过监测该突变的方法(ASO探针或酶切位点监测)来进行诊断。如果致病突变或病
关于染色体的小知识 一、什么是染色体? 染色体是一种由DNA、蛋白质及RNA等组成的核蛋白复合物,因易被碱性染料染色而得名。染色体是核基因的载体。高等生物都有染色体,但不同物种的染色体数目不相同。美国遗传学家Tjio和Levan在1956年首次发现人类正常体细胞染色体数目是46条(23对)。人类正常精子或卵子的染色体数目为23条。 二、染色体与性别决定 在人类体细胞的23对染色体中的22对都与性别无直接关系,称为常染色体。而另外1对与性别的决定有明显而直接关系的染色体——X染色体和Y染色体,称为性染色体。 正常女性细胞中有两条X染色体;而正常男性细胞中有一条X染色体和一条Y染色体。因此,女性只能产生含有X染色体的一种卵子;而男性则可以产生两种精子,即含X 染色体的X型精子和含Y染色体的Y型精子。受精时,X型精子与卵子结合形成含有两条X 染色体的受精卵,将发育成女性;Y型精子与卵子结合形成含有一条X染色体与一条Y染色
体的受精卵,将发育成男性。所以人类的性别是精子和卵子受精的瞬间决定的,确切的说是由精子决定的。 值得一提的是,X染色体与Y染色体在性别决定中的作用并不对等。Y染色体上有一个决定男性性别的基因——睾丸决定因子基因,是决定性别的关键基因。所以,一个个体的体细胞中无论有多少条X染色体,只要有Y染色体,通常都会表现为男性(两性畸形患者除外)。 三、染色体检查及分析技术 我中心目前采用外周血淋巴母细胞分裂中期染色体G显带技术,依照国际人类细胞遗传学会制定的ISCN标准,进行染色体核型分析。另外,我中心还即将从德国引进染色体自动化分析系统,以便更好地为各位患者服务。 四、染色体畸变 染色体畸变是指体细胞或生殖细胞内染色体发生异常的改变,包括数目畸变和结构畸变。导致染色体畸变的因素有多种,许多有害化学物质、放射线、病原体及病原体产生的
常见遗传病总结 常染色体显性遗传 软骨发育不全上臂、大腿短小畸形,腹部隆起;臀部后凸;身材矮小致病基因导致长骨两端软骨细胞形成出现障碍 常染色体隐性遗传 白化病患者皮肤、毛发、虹膜中缺乏黑色素,怕光,视力较差缺乏酪氨酸的正常基因,无法将酪氨酸转变成黑色素 先天性聋哑听不到声音,不能学说话,成为哑巴缺乏听觉正常的基因,听觉发育障碍 苯丙酮尿症智力低下缺乏苯丙氨酸羟化酶的正常基因,苯丙氨酸不能转化成酪氨酸而不能变成苯丙酮酸,中枢神经受损 X染色体显性遗传 抗维生素D佝偻病X型腿(O型),骨骼发育畸形,生长缓慢致病基因使钙磷吸收不良没,导致骨骼发育障碍 X染色体隐性遗传 红绿色盲不能分辨红色和绿色缺乏正常基因,不能合成正常视蛋白引起色盲
血友病受伤后流血不止缺乏凝血因子合成基因,导致凝血障碍 进行性肌营养不良患者肌无力或萎缩,行走困难正常基因缺乏,进行性肌肉发育障碍 染色体数目异常 常染色体21三体综合症智力低下,身体发育缓慢,面容特殊,眼间距宽,口常开,舌伸出第21号染色体多一条 性染色体性腺发育不良(XO)身材矮小,肘外翻,颈部皮肤松弛,外观女性无生育能力少一X染色体 XYY个体男性,身材高大,具有反社会行为多一Y染色体 八、人类几种遗传病及显隐性关系: 类别名称 单基因遗传病 常染色体遗传 隐性白化病、先天性聋哑、苯丙酮尿症 显性多指、并指、短指、软骨发育不全 性(X)染色体遗传 隐性红绿色盲、血友病、果蝇白眼、进行性肌营养不良
显性抗维生素D佝偻病 多基因遗传病唇裂、无脑儿、原发性高血压、青少年型糖尿病 染色体异常遗传病 常染色体病数目改变21三体综合症(先天愚型)结构改变猫叫综合症 性染色体病性腺发育不良 1、遗传基因的显隐性及在染色体上的位置: 常染色体隐性:先天性聋哑、苯丙酮尿症、镰刀型细胞贫血症、白化病。 常染色体显性:软骨发育不全、多指、并指、短指。 伴X隐性遗传:进行性肌营养不良、红绿色盲、血友病、果蝇白眼。 伴X显性遗传:果蝇红眼、抗VD性佝偻病、钟摆型眼球震颤。 伴Y遗传:鸭蹼病、人类印第安毛耳、外耳廓多毛症。 遗传病的判断是中学生物学考察的一个重要内容,掌握遗传病的判断方法和对常见遗传病进行归类是解决这类问题的重要手段。下面结合本人的教学实际并汇总K12网的部分内容,作如下说明: 一、遗传病的判断方法: 1、常染色体隐性遗传:一对正常的夫妇生出患病的女儿,一定是常染色体隐性遗传。说明:如果子代是儿子患病,只能说明该病是隐性遗传,该致病基因究竟是在常染色体上还是在性染色体上还需要进一步推理。 2、常染色体显性遗传:一对患病的夫妇生出正常的女儿,一定是常染色体显性遗传。说明:如果子代是儿子正常,只能说明该病是显性遗传,该致病基因究竟是在常染色体上还是在性染色体上还需要进一步推理。
常染色体显性遗传病常见的并发症有哪些,怎么根治! 常染色体显性遗传病常见的并发症 常染色体显性遗传病有什么并发症 一、并发病症 常染色体显性遗传病主要包括以下几种病征。 1、软骨发育不全其主要特征为四肢短小畸形,可能系遗传性侏儒症中最常见的类型。因长骨骺端软骨细胞形成障碍,影响骨的长度,但骨的宽度仍然增长,而导致四肢短小,侏儒体型。出生时即呈现四肢短而粗,躯干相对较长;手指短而粗,各指长度相仿,两手下垂不过髋关节;儿童期或成年后头部明显过大,前额突出,马鞍鼻,颏部大而前突。此外,尚有腰椎前凸或驼背,两下肢内弯,步态摇摆,X线检查长骨变短,弯曲,两端膨大,头颅和前盆均具特征。患者智力及生殖功能正常。女性患者妊娠后,因骨盆狭窄需剖宫分娩。本症为常染色体显性遗传,故子代中有半数发病机会,但有相当一部分患者为基因突变所致。有人认为此基因突变与父龄过高有关。若患者为纯合子,则有双倍的基因效应,常可致死。超声和X线检查可做产前诊断。微信:39健康百科(微信号:jibingbaike39),可了解该疾病更多治疗方法,发病原因,护理知识。 二、蜘蛛脚样指综合征 其特征为肢体过长,眼病和心血管异常。因长骨过度生长而呈身材细长体型,上下段比例失常,四肢长,尤其指、趾细长;肋骨异常呈漏斗胸;肌肉发育差,皮下脂肪少,关节松弛;晶状体异位或重度近视;主动脉瓣关闭不全或主动脉瘤。主动脉瘤破裂是早亡的主要原因,妊娠增加主动脉破裂的危险,尤其在分娩前后,虽无主动脉瘤,但妊娠后亦应作为高危妊娠监护。本征虽为常染色体显性遗传,但有时轻症者表现不典型而又无其他重症亲属可见时,则轻度患者无法确诊,咨询中发生困难。 3、多发性神经纤维瘤病本病为神经外胚层的病变,皮肤上有黄棕色的色素斑为典型的特殊体征,呈卵圆形或环状不一,直径为1~5cm;还常伴有皮肤神经纤维瘤,呈多发性,较小,质柔软,稀疏分布,大的神经纤维瘤常在外周神经或神经根上,可导致脊柱畸形。中枢神经系统的神经纤维瘤最常累及听神经,若有弥漫性病变则伴有轻度智力障碍。本病有伴发恶性肿瘤的危险,神经纤维瘤或其他部位均可并发恶性肿瘤,其几率为10%~20%。尚有脊柱侧凸、轻度智力障碍、癫痫、神经根压迫、胫骨假关节和嗜铬细胞瘤等。 三、遗传性舞蹈病 本病为大脑中基底核的病变,主要表现为进行性痴呆和不自主的舞蹈动作。起病隐匿,仅是正常的面部动作和手势增加,以后呈现舞蹈样的不随意运动,舞蹈样动作缓慢,两次动作的间歇期较长。早期诊断较为困难,虽能发现基底核的萎缩变化,但达不到早期诊断的要求。本病发病年龄在40 岁左右,少数可在儿童后期发病,早期即有智力减退,呈进行性痴呆,大多于10年后恶化。本病虽为常染色体显性遗传,但遗传咨询常达不到预期效果,因为患者大多于婚配且生育后发病,而其子女中虽有一半机会再患此病,但无法预测亦无法早期诊
1、一个体细胞中的全部染色体,按其大小、形态特征顺序排列所构成的图像就称为核型。 2、将待测细胞的核型进行染色体数目、形态特征的分析,确定其是否与正常核型完全一致,称为核型分析。 3、通过技术的改进从早中期、前中期、晚前期细胞得到更长、带纹更多的染色体。一套单倍体染色体即可显示550~850条或更多的带纹,这种染色体称为高分辨显带染色体。 4、在正常健康人群中,存在着各种染色体的恒定的微小变异,包括结构、带纹宽窄和着色强度等。这类恒定而微小的变异是按照孟德尔方式遗传的,通常没有明显的表型效应或病理学意义,称为染色体多态性。 5、在短胃和长臂的末端分别有一特化部位称为端粒。 6、人类近端着丝粒染色体的短臂末端有一球状结构,称为随体。 7、在某些染色体的长、短臂上还可见凹陷缩窄的部分,称为次级缢痕。 8、人体正常生殖细胞精子和卵子所包含的全部染色体称为一个染色体组。 9、随体柄部为缩窄的次级缢痕,次级缢痕与核仁的形成有关,称为核仁形成区或核仁组织区。 10、如果染色体的数日变化是单倍体(n)的整倍数,即以n为基数,整倍地增加或减少,则称为整倍体。 11、一个个体内同时存在两种或两种以上核型的细胞系,这种个体称嵌合体。 12、一条染色体的长、短臂同时发生了断裂,含有着丝粒的片段两断端发生重接,即形成环状染色体。 13、有时细胞中某些号的染色体数目发生了异常,其中有的增加,有的减少,而增加和减少的染色体数目相等,结果染色体总数不变,还是二倍体数(46条),但不是正常的二倍体核型,则称为假二倍体。 14、当体细胞中染色体数目减少了一条或数条时,称为亚二倍体。 15、是一个染色体上某一片段增加了一份以上的现象,使这些片一段的基因多了一份或几份。 16、两条染色体同时发生一次断裂后,两个具有着丝粒的片一段的断端相连接,形成了一条双着丝粒染色体。 17、是某一染色体发生两次断裂后,两断点之间的片段旋转180°后重接,造成染色体上基因顺序的重排。 18、一条染色体的两个臂在形态遗传结构上完全相同,称为等臂染色体。 19、体细胞内只含有单个染色体组称为单倍体。 20、一条染色体的断片一移接到另一条非同源染色体的臂上,这种结构畸变称为易位。 21、是两条染色体同时发生断裂,断片交换位置后重接形成的两条染色体。 22、两个近端着丝粒染色体在着丝粒部位或着丝粒附近部位发生断裂后,二者的长臂和短臂各形成一条新的染色体。 23、两条非同源染色体同时发生断裂,但只有其中一条染色体的片一段插入到另一条染色体的非末端部位。 24、是染色体片段的丢失,缺失使位上这个片段的基因也随之发生丢失。 25、体细胞内某对染色体少了一条,细胞染色体数日为45 (2n-1),称为单体型。 26、体细胞内某对染色体多了一条,细胞内染色体数目为47(2n+1),称为三体型。 27、体细胞内染色体多了两条或两条以上,即构成多体型。 28、体细胞内含有两个染色体组称为二倍体。 29、超过二倍体的整倍体被称为多倍体。 30、染色体数目或结构异常引起的疾病称为染色体病。 31、Turner综合征也称为女性先天性性腺发育不全或先天性卵巢发育不全综合征,为性染色
定义: 染色体病(chromosomal disorder): 染色体数目或结构异常引起的疾病 分类: 常染色体病、性染色体病、染色体异常的携带者 一、染色体的发病概况 染色体病在临床上和遗传上特点: 1.染色体病患者均有先天性多发畸形(包括特殊面容)、生长、智力或性发育落后、特殊肤纹。 2.绝大多数染色体病患者呈散发性,这类患者往往无家族史。 3.少数染色体结构畸变的患者是由表型正常的双亲遗传而得,这类患者常伴有家族史。 二、常染色体病(Autosomal disease) 1.定义: 由常染色体数目或结构异常引起的疾病 2.分类: 三体综合征、单体综合征、部分三体综合征、部分单体综合征、嵌合体等 (一)Down综合征(Down Syndrome,DS) 1.特点: 母亲生育年龄偏大和xx的一致性 2.发生率:
新生儿的DS发生率约为~;发生率随母亲生育年龄的增高而增高,尤其当母亲年龄大于35岁时,发生率明显增高。 3.表型特点: (1)是一种很明确的综合征 (2)多数情况下,都是新发生的、散在的病例 (3)同卵双生具有一致性 (4)男性患者没有生育力,而极少数女性患者可生育 (5)随母亲年龄增加该病的发生率也升高,尤其当母亲大于35岁时发病率明显升高 (6)患者免疫功能缺陷、易患先天性心脏病 (7)表型特征的表现度不同 (8)急性白血病死亡率增加了20倍 4.遗传分型 (1)游离型(Trisomy) ①游离型(21三体型)即标准型。此型约占全部患者的 92.5%。核型为47,XX(XY),+21。 ②此型的发生绝大部分与父母核型无关,它是生殖细胞形成过程中,在减数分裂时不分离的结果。染色体不分离发生在母方的病例约占95%,另5%见于父方,且主要为第一次减数分裂不分离。 (2)易位型(Robertsonian translocation) ①此型约占5%,增加的一条21号染色体并不独立存在,而是与D组或G 组的一条染色体发生罗伯逊易位,染色体总数为46,其中一条是易位染色体。
《染色体》 染色体(Chromosome )是细胞内具有遗传性质的物体,易被碱性染料染成深色,所以叫染色体(染色质);其本质是脱氧核甘酸,是细胞核内由核蛋白组成、能用碱性染料染色、有结构的线状体,是遗传物质基因的载体。 染色体 染色体是细胞核中载有遗传信息(基因)的物质,在显微镜下呈圆柱状或杆状,主要由脱氧核糖核酸和蛋白质组成,在细胞发生有丝分裂时期容易被碱性染料(例如龙胆紫和醋酸洋红)着色,因此而得名。 在无性繁殖物种中,生物体内所有细胞的染色体数目都一样;而在有性繁殖大部分物种中,生物体的体细胞染色体成对分布,称为二倍体。 性细胞如精子、卵子等是单倍体,染色体数目只是体细胞的一半。 [1]哺乳动物雄性个体细胞的性染色体对为XY,雌性则为XX。鸟类和蚕的性染色体与哺乳动物不同:雄性个体的是ZZ,雌性个体为ZW。 编辑本段发现 1879年德国生物学家弗莱明(F1eming·w )把细胞核中的丝状和粒状的物质,用染料染红,观察发现这些物质平时散漫地分布在细胞核中,当细胞分裂时,散漫的染色物体便浓缩,形成一定数目和一定形状的条状物,到分裂完成时,条状物又疏松为散漫状。 1883年美国学者提出了遗传基因在染色体上的学说。
染色体 1888年正式被命名为染色体。 1902年美国生物学家沃尔特·萨顿和鲍维里通过观察发现细胞的减数分裂时染色体与基因具有明显的平行关系,并推测基因位于染色体上。 1928年摩尔根通过果蝇杂交实验证实了染色体是基因的载体,从而获得了生理医学诺贝尔奖。 1953年4月《自然》杂志刊登了美国的沃森和英国的克里克在英国剑桥大学合作的研究成果:DNA双螺旋结构的分子模型,被誉为20世纪以来生物学方面最伟大的发现。 1956年,美籍华裔遗传学家Joe Hin Tjio (1919–2001,资料译为庄有兴或蒋有兴),和Levan 首次发现人的体细胞的染色体数目为46条,标志着人类细胞遗传学的建立。46条染色体按其大小、形态配成23对,第一对到第二十二对叫做常染色体,为男女共有,第二十三对是一对性染色体,雄性个体细胞的性染色体对为XY;雌性则为XX。 编辑本段结构 简介 染色体的超微结构显示染色体是由直径仅100埃(Å,1埃=0.1纳米)的DNA-组蛋白高度螺旋化的纤维所组成。每一条染色单体可看作一条双螺旋的DNA分子。有丝分裂间期时,DNA解螺旋而形成无限伸展的细丝,此时不易为染料所着色,光镜下呈无定形物质,称之为染色质。有丝分裂时DNA高度螺旋化而呈现特定的形态,此时易被碱性染料(例如龙胆紫和醋酸洋红)着色,称之为常染色体。 1970年后陆续问世的各种显带技术对染色体的识别作出了很大贡献。中期染色体经过DNA
遗传病遗传方式 多指(趾)常染色体显性遗传 并指(趾)常染色体显性遗传 蜘蛛指(趾)常染色体显性遗传 短指(趾)常染色体显性遗传 缺指(趾)常染色体显性遗传 先天性颅骨畸形常染色体显性遗传 下颌-面骨发育异常常染色体显性遗传 颅锁骨发育不全常染色体显性遗传 软骨发育不全常染色体显性遗传 多发性骨骺发育不全常染色体隐性遗传 成骨不全Ⅱ型常染色体显性遗传/常染色体隐性遗传 石骨症严重常染色体隐性遗传 轻型常染色体显性遗传 进行性肌营养不良 X连锁隐性遗传 强直性肌营养不良常染色体显性遗传/常染色体隐性遗传 先天性肌强直常染色体显性遗传/常染色体隐性遗传 家族性周期性麻痹常染色体显性遗传/常染色体隐性遗传 进行性骨化性肌炎常染色体显性遗传 支气管软化症常染色体隐性遗传 气管-支气管巨大症常染色体隐性遗传 不动纤毛综合症常染色体隐性遗传 肺泡性蛋白沉着症常染色体隐性遗传 肺囊性纤维症常染色体隐性遗传 弯刀综合症常染色体显性遗传 家族性气发性气胸常染色体显性遗传/常染色体隐性遗传 多发性家族**肉病常染色体显性遗传 色素沉着肠道息肉综合症常染色体显性遗传 先天性直肠肛门畸形常染色体显性遗传/常染色体隐性遗传α1-抗胰蛋白酶缺乏症常染色体隐性遗传 遗传性慢性再发性胰腺炎常染色体显性遗传 先天性心脏病常染色体显性遗传 房间隔缺损 室间隔缺损 动脉导管未闭常染色体显性遗传/常染色体隐性遗传 法洛四连症常染色体显性遗传 先天性颅骨畸形常染色体显性遗传 先天性肺动脉瘘常染色体显性遗传 家族性二尖瓣脱垂常染色体显性遗传 心内膜弹力纤维增生症常染色体隐性遗传 心-手综合症常染色体显性遗传 高脂蛋白血症Ⅰ型常染色体显性遗传
染色体病 定义:染色体病(chromosomal disorder):染色体数目或结构异常引起的疾病 分类:常染色体病、性染色体病、染色体异常的携带者 一、染色体的发病概况 染色体病在临床上和遗传上特点: 1.染色体病患者均有先天性多发畸形(包括特殊面容)、生长、智力或性发育落后、特殊肤纹。 2.绝大多数染色体病患者呈散发性,这类患者往往无家族史。 3.少数染色体结构畸变的患者是由表型正常的双亲遗传而得,这类患者常伴有家族史。 二、常染色体病(Autosomal disease) 1.定义:由常染色体数目或结构异常引起的疾病 2.分类:三体综合征、单体综合征、部分三体综合征、部分单体综合征、嵌合体等 (一)Down综合征(Down Syndrome,DS) 1.特点:母亲生育年龄偏大和单卵双生的一致性 2.发生率:新生儿的DS发生率约为1/1000~2/1000 ;发生率随母亲生育年龄的增高而增高,尤其当母亲 年龄大于35岁时,发生率明显增高。 3.表型特点:(1)是一种很明确的综合征 (2)多数情况下,都是新发生的、散在的病例 (3)同卵双生具有一致性 (4)男性患者没有生育力,而极少数女性患者可生育 (5)随母亲年龄增加该病的发生率也升高,尤其当母亲大于35岁时发病率明显升高 (6)患者免疫功能缺陷、易患先天性心脏病 (7)表型特征的表现度不同 (8)急性白血病死亡率增加了20倍 4.遗传分型 (1)游离型(Trisomy) ①游离型(21三体型)即标准型。此型约占全部患者的92.5%。核型为47,XX(XY),+21。 ②此型的发生绝大部分与父母核型无关,它是生殖细胞形成过程中,在减数分裂时不分离的结果。染 色体不分离发生在母方的病例约占95%,另5%见于父方,且主要为第一次减数分裂不分离。(2)易位型(Robertsonian translocation) ①此型约占5%,增加的一条21号染色体并不独立存在,而是与D组或G组的一条染色体发生罗伯 逊易位,染色体总数为46,其中一条是易位染色体。 ②最常见的是D/G易位,如核型为46,XX(XY),-14,+t(14q21q),其次为G/G易位,如核型为46, XX(XY),-21,+t(21q21q)。 (3)嵌合型(Mosaicism) 此型较少见,约占2%。此型产生的原因:一是由于生殖细胞减数分裂不分离,继而因分裂后期染色体行动迟缓引起部分细胞超数的染色体发生丢失而形成含有47,+21/46两个细胞系的嵌合体,由此形成的嵌合体的发生率与标准的三体型一样,随母亲年龄的增加而增加。二是合子后(post-zygotic)有丝分裂不分离的结果。 5.分子机制 (1)21号染色体的分子解剖学 (2)21号染色体上与DS表型相关的基因 ①与智力发育迟缓相关的基因 DS细胞粘附分子DSCAM基因:编码一种细胞粘附分子,表达在成人脑组织中,参与神经系统分化,与DS中枢和外周神经缺陷有关。 活性依赖性神经保护蛋白ADNP基因:在海马、大脑皮质和小脑中表达,是一新型的热休克蛋白,