常染色体显性遗传性共济失调
- 格式:ppt
- 大小:215.00 KB
- 文档页数:35

在肌力没有减退的情况下,肢体运动的协调动作失调,不平稳与不协调,称为共济失调(ataxia)。
【病因和机理】机体任何一个简单的运动必须有主动肌、对抗肌、协同肌、固定肌四组肌肉的参与才能完成,并有赖于神经系统的协调与平衡。
脊髓后索的薄束与楔束贯穿脊髓之全长,薄束传导躯干下段与两下肢的深感觉,楔束传导躯干上段与两上肢的深感觉。
从后索发出的纤维在延髓交叉,经对介的丘脑而到大脑皮质,后束传导肌肉、关节与肌腱的深感觉、肢体在空间中的位置、肢体运动的力与范围的冲动、以及部分感觉与两点鉴别感觉。
前旋系统向心传导平衡信息,引起平衡反应。
小脑是维持躯体平衡、共济运动和肌张力的重要中枢。
这些结构的功能又都是在大脑皮质的统一控制下完成的。
深感觉、前旋系统、大脑和上脑的病损均可发生共济失调,分别称为感觉性、前旋性、小脑性和大脑性共济失调。
【临床表现】1.感觉性共济失调:共济失调在睁眼时减轻,闭目时加剧,伴有位置觉,震动觉减低或消失。
因深感觉障碍下肢重而多见,故站立不稳和步态不稳为主要表现。
患者夜间行路困难,洗脸时躯体容易向脸盆方向倾倒(洗脸盆征阳性)。
行走时双目注视地面举足过高,步距宽大,踏地过重,状如跨阈,故称跨阈步态。
闭目难立征阳性,指鼻试验,跟膝胫试验不正确。
2.小脑性共济失调:小脑及其传入传出纤维病变都可引起共济失调,特点是既有躯干的平衡障碍而致站立不稳,也有肢体的共济失调而辨距不良、轮替运动障碍、协调不能、运动起始及终止延迟或连续性障碍。
小脑性共济失调不受睁眼、闭目或照明度影响,不伴感觉障碍,有眼球震颤、构音障碍、讷吃和特殊小脑步态,即行走时两足分开,步距大小不一,步态蹒跚不稳易倾倒。
指鼻试验时共济失调极为明显,可见上肢呈弧形摆动与意向性震颤,并有肌张力减低或消失、关节运动过度、快复动作障碍、肌肉反跳现象。
3.前庭性共济失调:因前庭系统损害引起,以平衡障碍为主。
特征为静止与运动时均出现平衡障碍。
与小脑性共济失调有相同点,如站立时两足基底宽、身体不稳、向侧方或后方倾倒、步行时偏斜等。


1例常染色体隐性遗传脊髓小脑共济失调1型的临床特点及基因分析杨丽1,马子珊1,马伯年1,罗嘉嘉1,张维1,杨智峰1,任紫晗1,兰甜甜1,陈桂生2,3摘要:本研究对1例因SETX基因突变引起常染色体隐性遗传脊髓小脑共济失调1型(SCAR1)的患者进行了临床表型分析及基因检测。
通过病史采集、神经系统查体、影像学检查、神经电生理检查及基因学分析,发现该患者9号染色体上的SETX基因存在复合杂合突变:c.5812C>T (p.Q1938X)和c.501_508del,这些突变位点分别位于第14外显子和第6外显子,且此前尚未在文献中报道。
本研究首次发现这两个突变可引起SCAR1,为进一步理解SETX基因在SCAR1中的致病机制提供了新的线索,并可能为未来类似病例的诊断和治疗提供参考。
关键词:常染色体隐性脊髓小脑共济失调1型;共济失调伴眼动失用2型;AOA2;SETX;复合杂合突变中图分类号:R744 文献标识码:AClinical and genetic features of autosomal recessive spinocerebellar ataxia type 1:A case report YANG Li,MA Zishan,MA Bonian,et al.(The First Clinical Medical College of Ningxia Medical University,Yinchuan 750004,China)Abstract:This study analyzes the clinical phenotype and genetic testing results of a patient with autosomal recessive spinocerebellar ataxia type 1 (SCAR1) caused by SETX gene mutations. Through medical history collection, neurological examination, radiological examination, neural electrophysiological examination, and genetic analysis, compound heterozy⁃gous mutations were found in the SETX gene on chromosome 9, i.e.,c.5812C>T (p.Q1938X) and c.501_508del, and these mutation sites were located in exon 14 and exon 6, respectively, and had not been reported in the literature. This study discovers for the first time that these two mutations can cause SCAR1, providing new insights into the pathogenic mechanism of the SETX gene in SCAR1 and a reference for the diagnosis and treatment of similar cases in the future.Key words:Autosomal recessive spinocerebellar ataxia type 1;Ataxia with oculomotor apraxia type 2;AOA2;SETX;Compound heterozygous mutation常染色体隐性遗传脊髓小脑共济失调1型(au⁃tosomal recessive spinocerebellar ataxia type 1,SCAR1),又称共济失调伴眼动失用2型(ataxia with oculomotor apraxia type 2,AOA2),是一种以早发型进行性小脑共济失调、多发性神经病变和甲胎蛋白水平升高为特征的神经系统变性疾病[1]。

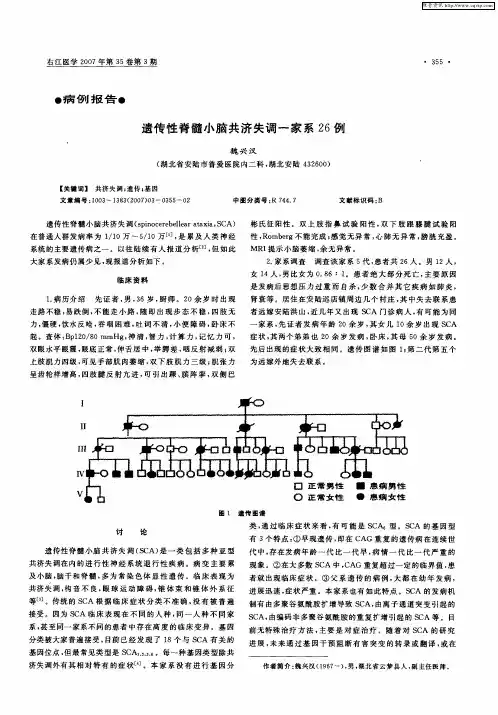


•论著.脊髓小脑性共济失调1型一哈萨克族家系遗传学特征分析承马建华F雷晶*张艳”【摘要】目的探讨脊髓小脑性共济失调1型(spinocerebellar ataxia type1,SCA1)一哈萨克族家系遗传学特征。
方法应用聚合酶链反应、琼脂糖凝胶电泳、T载体连接克隆测序等技术对42例家系成员进行ATXN1基因CAG三核昔酸重复数目测定。
对检测结果进行分析并与表型正常的家系成员和健康人对照。
结果42例家系成员中SCA1基因突变者19例,5例已发病SCA1患者异常等位基因CAG重复数目为41~47次。
家系内表型正常的23例成员SCA1等位基因CAG重复数目为17-30次。
60名健康对照者SCA1等位基因CAG重复数目为12-30次。
在正常等位基因中发现CAT结构的插入。
结论哈萨克族SCA1家系是国内首次报告少数民族SCA1家系,CAG异常扩增数目在不同民族中存在差异。
等位基因中CAT结构的插入更有利于基因的稳定性。
【关键词】脊髓小脑性共济失调1型哈萨克族三核昔酸重复ATXN1基因CAT结构【中图分类号】R744.7【文献标识码】AThe analysis of genetic characteristics of spinocerebellar ataxia type1in a Kazak family.MA Jianhua,LEI Jing,ZHANG Yan.Depatment of Neurologu,,y>the First Affilaied Hospital of Xinjiang Medical University,Urumqi 830054,China.Tel:0991-*******.[Abstract]Objective To explore the genetic characteristics of the spinocerebellar ataxia type1(SC Al)in a Kazakh family.Methods Genetic test(ATXN1gene)of42family members were conducted by polymerase chain reaction,agarose gel electrophoresis and T vector ligand sequencing.Genetic test results were analyzed and comparisonof phenotypes were made between family members and healthy people.Results Among the42family members,19hadthe mutation of SC A l gene,and the number of CAG repeats of the abnonnal allele in5patients with SC A l was41to47, while the number of CAG repeats in the23members with nonnal phenotype in the family was17to30.The number ofthe SC A l allele CAG in60healthy controls were12to30.Insertion of CAT structure found in normal alleles. Conclusion The Kazak SC A l family is the first reported of SC A l in ethnic minority families in China,and the abnormal length of CAG repeats varies among different ethnic groups.The insertion of the CAT structure in the allele tends to increase the stability of the gene.[Key words]Spinocerebeller ataxia type1Kazakh family Triple nucleotide repeats ATXN1gene CAT structure脊髓小脑性共济失调1型(spinocerebellar ataxia type1,SC A l)是一种常染色体显性遗传性神经系统退行性疾病,通常在30〜40岁发病。

共济失调共济失调(ataxia)是指肌力正常的情况下运动的协调障碍。
肢体随意运动的幅度及协调发生紊乱,以及不能维持躯体姿势和平衡。
但不包括肢体轻度瘫痪时出现的协调障碍、眼肌麻痹所致的随意运动偏斜,视觉障碍所致的随意运动困难以及大脑病变引起的失用症。
任何一个简单的运动必须有主动肌、对抗肌、协同肌和固定肌四组肌肉的参与才能完成,并有赖于神经系统的协调和平衡。
共济失调的病因很多,深感觉(深感觉是指感受肌肉、肌腱、关节和韧带等深部结构的本体感觉。
肌肉是处于收缩或舒张状态;肌腱和韧带是否被牵拉以及关节是处于屈曲还是伸直的状态等的感觉。
检查方法①振动觉检查:置振动的128Hz音叉末端于骨突起处(例如内外踝、膝盖、髂前上棘、腕骨或脊椎棘突等处)以试验患者能否察觉。
注意感受的时限,两侧对比。
②位置觉检查:瞩患者闭目,移动患者一肢的大多数关节,塑成一种姿势,瞩患者保持之,然后瞩患者用对侧的一肢模仿。
③运动觉检查:轻移患者的手指和足趾向上及向下,瞩患者说出移动的方向)、前庭系统、小脑和大脑损害都可发生共济失调,根据病变部位不同,共济失调可分为四种类型:①深感觉障碍性共济失调;②前庭迷路性共济失调;③小脑性共济失调;④大脑型共济失调。
而一般称呼的“共济失调”,多特指小脑性共济失调。
几乎100%的Ias(颅内动脉粥样硬化性狭窄)患者有共济失调的表现。
还有原因不明的因素,有的伴有智能不全或痴呆。
神经系统的协调和平衡包括:1.感觉性深感觉向中枢神经系统反映躯体各部位的位置和运动方向。
病因有:①周围神经或神经根病;②脊髓亚急性联合变性(简称亚急性联合变性(subacute combined degeneration,SCD),是由于维生素B12的摄入、吸收、结合、转运或代谢障碍导致体内含量不足而引起的中枢和周围神经系统变性的疾病。
病变主要累及脊髓后索、侧索及周围神经等,临床表现为双下肢深感觉缺失、感觉性共济失调、痉挛性瘫痪及周围性神经病变等,常伴有贫血的临床征象。

一、概述下列哪些是单基因遗传病的遗传方式( )提交并继续图 1.2 系谱图二、常染色体显性遗传病如果疾病的致病基因位于第 1 到 22 号染色体上,其遗传方式是显性的,即杂合时可以发病,这种疾病称为常染色体显性遗传病。
其常见婚配类型是一个患常染色体显性遗传病的患者和一个表型正常的人结婚,其后代会有 50% 遗传到致病基因,50% 遗传到正常基因的染色体(图 2.1 );另一种情况是常染色体显性遗传新生突变,即两个正常的人婚配,生出患病的子代(图 2.2 )。
(一)常染色体显性遗传的系谱特征合子,只是出现根腱部的腱黄瘤(图 2.8 ),症状比患者轻,血胆固醇也比患者(纯合型)低,由于症状较轻,发生动脉粥样硬化、冠心病的时间会相应的晚一些(二三十岁、三四十岁)。
( 2 )短肢侏儒症另外较常见的是短肢侏儒,此类病人大多为软骨发育不全,其特征是头部较大,前额突出,面中部发育不良,躯干相对较长,同时有 O 型腿(膝内翻),手指伸开后呈车轮状或者称三叉手;同时患者的腰椎明显前突(图 2.9 )。
其基因改变是明显的杂合改变,此类患者是可以生存的。
图 2.9 软骨发育不全的杂合表现图 2.10 是软骨发育不全的纯合改变,此类患者骨骼严重畸形,在宫内时胸廓很小,所以因为呼吸窘图 2.11 三节拇指并多指患者图 图 2.12 三节拇指并多指患者母亲图 2.13 三节拇指并多指系谱图图 2.14 是 A 型轴后多指的系谱,四代出现 1 个患者(图 2.15 ),患者的父亲母亲无任何表型,但子代出现患者,说明其是不外显患者。
轴后多指的额外指在小指一侧,A 型的额外指发育良好,与第 5指形成关节,外显率可达 75% ;B 型的额外指发育不良,常常只形成一个皮肤赘,外显率为 65% 。
图 2.14 A 型轴后多指的系谱图 图 2.15 A 型轴后多指患者( 2 )表现度导致不规则显性的另一个原因是表现度。
表现度是指致病基因的表达程度,可以有轻度、中度和重度的不同,称为可变的表现度。
企鹅病怎么复健治疗
企鹅病可能大家都比较陌生,不怎么听说过,它就是指遗传性小脑性共济失调,是所有报道的共济失调中最多的一种,患者行走如企鹅般摇摇晃晃,遗传方式为常染色体显性遗传,男女发病率无明显差异。
那么你们知道企鹅病怎么复健治疗吗?一起来看看吧。
企鹅病的临床表现
1、多在30~60岁起病,少数在少年期或70岁时发病。
有常染色体显性遗传的家族史,常以共济失调步态为首发症状,行走不稳,易跌倒,此后可逐渐出现双上肢共济失调。
2、部分病例可有吞咽困难、失音、言语障碍、情绪不稳、智慧衰退等,也可有复视、眼球活动障碍等。
3、双下肢无力,肌张力增高,腱反射亢进或减退,可有病理反射阳性。
4、无弓形足及心脏异常。
企鹅病的诊断鉴别
1、发病年龄较迟,有常染色体显性遗传的家族史。
2、进行性加重的共济失调,步态为醉酒样。
3、无心脏异常及弓形足,骨骼X线照片常正常。
4、排除其他类型的共济失调,排除脑瘫和运动神经元疾病及癌性小脑共济失调。
企鹅病的复健治疗
1、尽量保持与社会接触,争取生活平衡。
2、选择适合自己的工作和生活方式,尽可能与别人多交往,保持愉悦的心理状态。
3、选择适合自己体能状态的运动,以维持心肺耐力、肌力,使身体的柔软度处于最佳状态。
4、注意生活起居,不要固定在相同的姿势太久,常常活动手脚。
5、接受物理治疗、职业治疗或言语治疗,用以舒缓病情。
6、亲人的爱心照顾可增强患者的生命力。