成人多囊肾PKD1和PKD2
- 格式:ppt
- 大小:1.30 MB
- 文档页数:27

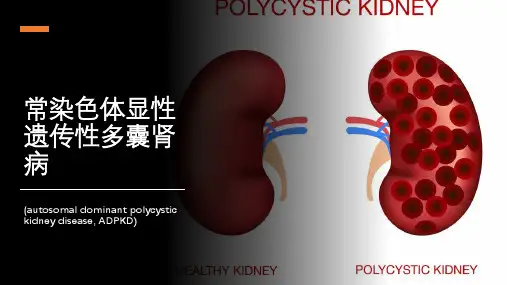
【案例分享】⼀例多囊肾的基因检测概述多囊肾病(PKD)是⼈类常见的单基因遗传病之⼀,按遗传⽅式可分为常染⾊体显性多囊肾病(ADPKD)和常染⾊体隐性多囊肾病(ARPKD)。
常染⾊体隐性多囊肾,是幼⼉型多囊肾,发病于婴⼉期,临床较罕见,发病率1/10000-1/40000;常染⾊体显性多囊肾,是成⼈型多囊肾,常于青中年时期被发现,也可在任何年龄发病,为多囊肾的常见类型,发病率为1/400-1/1000。
多囊肾病(PKD)的特征是肾中液体囊肿。
这是慢性肾功能不全和终末期肾病的第四⼤常见原因。
除累及肾脏外,常伴肾外囊肿,如肝囊肿、胰腺囊肿、脾囊肿等。
常染⾊体显性多囊肾病(ADPKD)是最常见的危及⽣命的遗传性疾病,其特征在于囊肿形成及肾脏和其他器官的肿⼤。
在ADPKD中有两种已知的基因突变:PKD1基因和PKD2基因。
PKD1是编码多囊蛋⽩1的⼤⽽复杂的基因,PKD2较⼩,编码多囊蛋⽩2。
ADPKD与其他囊性肾病的鉴别诊断取决于患者的年龄,家族史和相关表现。
在没有ADPKD家族史的成⼈患者中,医⽣应排除常染⾊体隐性多囊肾病(ARPKD)。
幼⼉在没有ADPKD家族史的情况下,重要的是区分ARPKD。
常染⾊体隐性多囊肾病(ARPKD)通常⼜被成为婴⼉性多囊肾病,属于肝肾纤维囊性疾病,典型者在婴⼉期发病,同时也是⼉科终末期肾脏病的重要原因之⼀,⼈群携带者为1:70。
30.5-50%的患病胎⼉因⽺⽔过少导致肺发育不全⽽在围产期死亡,此期主要为肾损害。
影像学检查主要表现为双肾集合管的⾮梗阻性梭形膨⼤,回声超强,呈陶⼟样“Potter”⽺⽔过少症。
如果度过新⽣⼉期,⼀般患⼉会进展⾄终末期肾病。
症状体征ADPKD主要表现为双侧肾囊肿且逐渐发展,肾脏体积进⾏性增⼤,肾⼩球滤过率逐步降低,常出现⾼⾎压(60%)、腹痛(61%)、⾎尿(15%)、蛋⽩尿(11%)、囊肿或尿路感染、肾结⽯(10%-35%)等并发症。
此外,ADPKD属多系统性疾病,35岁以上患者中超过90%合并肝囊肿、颅内动脉瘤、胰腺囊肿、结肠憩室或⼼脏瓣膜异常;其中肝囊肿是最常见合并症(83%-94%)。



胡桃夹综合征(nutcracker phenomenon)即(左肾静脉压迫综合征),又称胡桃夹现象,好发于青春期至40岁左右的男性,儿童发病分布在4~7岁,多发年龄见于13~16岁。
人体的血管像四通八达的道路一样,是有一定走向的。
左肾静脉行走在腹主动脉和肠系膜上动脉之间,这两条动脉构成40~60度的夹角,左肾静脉刚好通过此夹角。
从解剖上看,右肾静脉径直注入下腔静脉,行程短而直。
而左肾静脉则需穿过腹主动脉和肠系膜上动脉之间的夹角,跨越腹主动脉前方始能注入下腔静脉,因此左肾静脉远较右肾静脉长。
正常时,肠系膜上动脉与腹主动脉之间的夹角被肠系膜、脂肪、淋巴结和腹膜等所充塞,使左肾静脉不致受到压挤。
当青春期发育较快、身高迅速增长、脊柱过度伸展、体形急剧变化或肾下垂等情况下,左肾静脉在这个夹角中的日子就不好过了,会受到挤压,引起血流变化和相应的临床症状。
症状:胡桃夹现象的主要症状是血尿和蛋白尿,其中无症状肉眼血尿更易发现。
血尿的原因是左肾静脉受压致肾静脉高压,左肾静脉扩张所引流的输尿管周围静脉与生殖静脉淤血、与肾集合系统发生异常交通,或部分静脉壁变薄破裂,引起非肾小球性血尿,还会发生睾丸静脉和卵巢静脉淤血而出现肋腹痛,并于立位或行走时加重。
另外男性还能发生精索静脉曲张。
此外有蛋白尿,不规则月经出血,高血压等。
此病的诊断标准为:一侧肾出血;尿红细胞形态为非肾小球性;尿中钙排泄量正常;膀胱镜检查为左侧输尿管口喷血或血性尿;腹部彩超或CT检查可见左肾静脉扩张等。
超声检查:超声对胡桃夹综合征的诊断有着明显的优势,超声检查时可清晰显示腹主动脉、肠系膜上动脉及左肾静脉的解剖情况,在不同横断面均可找到左肾静脉扩张近段的最大内径,测值准确,同时可观察并测量肠系膜上动脉与腹主动脉夹角变化。
彩超血流速度提供更准确的血流动力学变化,有助于此病诊断。
超声检查还能除外先天性畸形、外伤、肿瘤、结石、感染性疾病及血管异常等造成的血尿。
多囊肾多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。

常染色体显性多囊肾(Autosomal dominant polycystic kidney disease,ADPKD)又称为成人型多囊肾,是一种遗传性全身性疾病,主要影响肾脏,但也可能会影响其他器官,如肝脏、胰腺、脑动脉血管等。
患有这种疾病的人大约有一半将会发展为终末期肾脏疾病,需要进行透析或肾移植。
患者进展为终末期肾病通常发生在40-60岁之间[1]。
常染色体显性多囊肾病在全球范围都有发生,发病率大约为1/400人-1/1000人[2][3]。
现在认为,常染色体显性多囊肾和两个基因缺陷有关。
85%的患者是由位于16号染色体的基因PKD1(TRPP1)发生突变所致,15%的患者是由PKD2(TRPP2)突变所致[1]。
常染色体显性多囊肾需要和常染色体隐性多囊肾进行辨别。
常染色体隐性多囊肾也导致肾脏和肝脏的囊肿,但通常只发生在童年,发病率约为1/20000人[4],病因和预后都和常染色体显性多囊不同。
目录 [隐藏]1 病理生理学2 诊断3 治疗4 预后5 参考资料6 外部链接病理生理学 [编辑]近期应用实验模式生物如线虫(秀丽隐杆线虫)和家鼠的纤毛和鞭毛进行基本细胞生物学研究使人类发生常染色体显性多囊肾的原因变得清楚。
生化学家James Calvet写到:“这些发现证明了遗传学和动物模型的能力和重要性。
如果没有这些遗传学研究的指引,有谁会想到一条小小的纤毛会成为研究多囊肾病的基础?”[5]纤毛在肾脏发育中发挥重要作用,纤毛结构功能异常直接导致肾囊肿性疾病的发生。
所有的纤毛和鞭毛的装配和维持都需要依赖鞭毛内运输这个非常重要的生理过程来完成。
鞭毛内运输是蛋白质插入纤毛和鞭毛膜特定位点必不可少的一项细胞功能。
这些插入的膜蛋白可以启动环境反馈和细胞内信号转导通道。
它们在肾小管上皮细胞的纤毛中发挥特殊作用,通过上述鞭毛内运输机制定位于肾小管上皮细胞的纤毛,被认为是正常肾细胞的发育和发挥功能的关键。
纤毛上皮细胞排列于尿收集管的内腔,感觉尿流率的变化。


常染色体显性遗传多囊肾研究进展摘要常染色体显性遗传多囊肾(ADPKD)是一种发病率高、预后差的疾病,它在发病机制、治疗等方面有很多进展。
关键词常染色体显性遗传多囊肾;瞬时受体势;Max作用因子1;雷帕霉素靶蛋白常染色体显性遗传多囊肾(autosomal dominant polycystic kidney disease,ADPKD)是一种常见的遗传性肾病,是导致肾衰竭的重要疾病。
现在已经发现3个基因(PKD1、PKD2、PKD3)与此病有关,其中PKD1定位于染色体16p13.3,其突变而导致ADPKD约占85%,PKD2定位于染色体4q21-23,其突变约占15%。
PKD3突变仅在几个家族中发现,目前尚未定位[1]。
ADPKD病变以双肾多发性进行性充液囊泡为主要特征。
囊泡损伤肾组织,引起肾功能改变,出现血尿、蛋白尿等临床症状,最终导致肾衰竭。
ADPKD除累及肾脏外,还可引起肝脏囊肿、胰腺囊肿、心脏瓣膜病、结肠憩室和颅内动脉瘤等肾外病变[2],给患者、家属及社会带来沉重负担。
故揭示其发病机理,研究新的治疗方法有很重要的意义。
本文对这些进展作以综述。
1 ADPKD的发病机制1. 1 多囊蛋白PC PKD1基因的蛋白产物被称为多囊蛋白-1(polycystin-1,PC1),又叫做TRPP1,多囊蛋白-1是一种跨膜蛋白,分布广泛,可以与多种蛋白(如PC2)、糖、脂类结合并发生交互作用,从而发挥功能。
PC1还与Wnt信号途径、JAK-STAT途径、转录因子AP-1和G蛋白偶联等信号途径有关。
PKD2基因的蛋白产物被称为多囊蛋白-2(polycystin-2,PC2),又叫做TRPP2,是TRP家族的一员,为非选择性钙离子通道。
它不同于PKD1,在人类基因组中是单拷贝,并不含多嘧啶区,但它的第一个外显子富含GC,从而使得此处易于突变[3]。
瞬时受体势(transient receptor potential,TRP)通道虽然最初发现于感受器,主要参与神经传导,但随着研究的深入,近年来人们发现该通道在肾脏病的发生、发展中也发挥了作用。

常染色体显性遗传性多囊肾病发病机制、影像学表现、分子诊断、临床表现和对症处理常染色体显性遗传性多囊肾病 (ADPKD) 是多囊肾病的一个亚型,为最常见的单基因遗传肾病,人群发病率为 1/1000~1/400。
ADPKD 常在40~70 岁期间进展为终末期肾病(ESRD),约占 ESRD 病例的 4.7%。
ADPKD 是一种全身性疾病,以双肾和身体其他部位出现大量液性囊泡为主要特征,可引起多种临床表现。
发病机制ADPKD 具有遗传异质性,位于 16 号染色体上的PKD1 基因(见于85% 的病例)或位于 4 号染色体上的PKD2 基因(见于 15% 的病例)突变与发病密切相关。
PKD1 和 PKD2 基因分别编码完整的膜蛋白多囊蛋白-1 和多囊蛋白-2,两者结构相似,可发生相互作用。
PKD1 或 PKD2 的突变可导致信号失调,环磷酸腺苷水平升高,最终导致囊肿生成。
鉴于 ADPKD 的显性遗传特性,患病父母的后代的发病率 50%。
5% ADPKD 由自发突变引起。
健康肾小管代偿性超滤的作用,肾脏囊肿形成和扩张并不引起明显的肾单位丢失,肾小球滤过率(eGFR)常数十年维持在正常水平。
绝大多数患者在 40 岁以后才开始出现肾功能不全,随后肾小球滤过率平均每年下降 4.4~5.9 mL/min。
影像学诊断ADPKD 的诊断需要从多方面综合诊断,主要考虑两点:①有无家族史,父母是否患有此病;②有无形态典型的多发肾囊肿,可通过影像学检查(CT、MRI、B 超)发现。
超成本较低、不需使用造影剂和无辐射暴露,是首选的筛查方式。
CT 和 MRI 在筛查超声无法诊断的病态肥胖患者中发挥作用。
分子诊断对于那些缺乏家族史或影像学检查无法确诊的多囊性肾病(PKD)患者,分子诊断显得尤为重要。
通过分析患者是否存在PKD1及PKD2基因突变,可以明确疾病诊断。
目前主要的技术手段包括基因连锁分析、直接检测基因突变、单链构象多态性分析及变性高效液相色谱(DHPLC)等。

多囊肾的遗传性及早期治疗创新多囊肾是一种常见的遗传性疾病,它导致肾脏内出现多个囊肿。
这些囊肿会逐渐增大并且影响到正常的肾功能。
本文将探讨多囊肾的遗传性以及早期治疗方面的创新进展。
一、多囊肾的遗传性1. 遗传方式:多囊肾可以通过两种方式遗传给下一代。
其中,ADPKD(成人型多囊肾)为最常见的类型,约占所有多囊肾患者的90%以上。
ADPKD是由一个突变基因造成,它可以从父母亲任意一个有此基因变异的人遗传给子女。
另外,ARPKD(婴儿型多囊肾)也是一种较为罕见但严重的遗传疾病,在这种情况下,需要双亲都带有该突变基因才能出现该病。
2. 基因突变:对于ADPKD患者而言,突变基因往往位于染色体16和染色体4上,并分别编码了称为PKD1和PKD2的蛋白质。
这些蛋白质在细胞中的功能紊乱使得囊肿在肾脏内增长。
此外,一些罕见的突变也与多囊肾相关,如PKHD1、GTPBP2和HNF1B基因的突变与ARPKD有关。
3. 遗传咨询:对于有家族性多囊肾病史的人群,建议进行遗传咨询。
遗传咨询可以帮助患者理解疾病的风险、遗传方式以及治疗选择。
此外,通过遗传测试可以了解个体患病风险,从而进行早期干预和治疗。
二、多囊肾早期治疗的创新进展1. 干扰物质治疗:近年来,一些药物被用于尝试延缓多囊肾发展。
其中最有希望的是在美国和欧洲获批上市的Tolvaptan。
Tolvaptan属于一种利尿剂,通过减少囊肿内液体分泌来抑制囊肿生长速度,并且能够改善患者的血压控制。
2. 基因编辑技术:基因编辑技术开辟了新的可能性,它可用于修复多囊肾相关基因的突变。
通过CRISPR-Cas9技术,研究人员可以针对特定的基因序列进行编辑,纠正突变并恢复正常功能。
然而,这项技术目前仍处在实验室阶段,并且还需要进一步研究确保其安全性和有效性。
3. 干细胞治疗:干细胞治疗是一种有望改善多囊肾患者肾功能的创新方法。
通过将干细胞移植到患者体内,这些干细胞可以分化为健康的肾组织,取代受囊肿侵蚀的肾单位。

中国人成人型多囊肾病PKD1基因突变筛选张兰;朱晓峰;李黎;罗建红【期刊名称】《解放军医学杂志》【年(卷),期】2000(025)006【摘要】@@ 成人型多囊肾病(Adult polycystic kidney dicease,APKD)是人类常见的常染色体显性遗传疾病,是8%~10%终末肾功能衰竭的病因.已知有3个基因的突变可导致APKD,其中PKD1和PKD2已被克隆和测序[1、2] ,PKD3也已定位[3].85%APKD是因PKD1基因突变所致,国外已发现近50种不同的突变方式[4].国内朱世乐等曾用与PKD1位点紧密连锁的微卫星DNA遗传标记,对中国人APKD家系成员作了单倍体型分析[5].本研究采用聚合酶链反应-单链构象多态性分析法(PCR-SSCP), 筛选分析了中国人APKD病人PKD1基因的突变.【总页数】2页(P436-437)【作者】张兰;朱晓峰;李黎;罗建红【作者单位】310013,杭州,解放军第117医院;310013,杭州,解放军第117医院;310013,杭州,解放军第117医院;浙江大学医学院医学分子生物实验室【正文语种】中文【中图分类】R6【相关文献】1.PKD1基因新突变导致常染色体显性多囊肾病 [J], 张慧峰;于为民;郭永艳;吕艳;尚雁;薛晋杰2.与PKD1基因连锁的微卫星DNA多态性及在成人型多囊肾病中的应用 [J], 赵铖3.PKD1,Polycystin与成人型多囊肾病 [J], 石蓉4.靶向序列捕获联合高通量测序鉴定成人型多囊肾PKD1基因新突变 [J], 沙艳伟5.与成人型多囊肾病PKD1基因紧密连锁的三种微卫星DNA在维吾尔族人群中的多态性研究 [J], 周勇;赵荣枝;沙莎;米力克扎提.巴吾东;郭卉因版权原因,仅展示原文概要,查看原文内容请购买。