利用二代测序技术检出成人型多囊肾PKD1基因新突变
- 格式:pdf
- 大小:136.23 KB
- 文档页数:2

医学机能实验学综述论文题目:多囊肾的分子遗传、发病机制及治疗的研究进展班级:2010级口腔班姓名:闫子玉学号:2010508060262012年12 月 5 日多囊肾的分子遗传、发病机制及治疗的研究进展闫子玉综述(青岛大学医学院,2010级本科生)摘要:多囊肾是最常见的常染色体遗传病,有约50%最终发展为终末期肾功能衰竭。
近年来,该疾病的主要基因PKD1和PKD2的克隆测序陆续完成,对PKD的基因结构、分子发病机制以及治疗的研究取得了很大的确进展,本文主要对这些进展作以综述。
关键词:多囊肾病分子遗传学发病机制治疗研究进展引言:多囊肾病(polycystic kidney disease,PKD)是指双侧肾脏发生多个囊肿且进行性增大进而导致肾脏结构和功能损害的一种最常见的常染色体遗传性疾病。
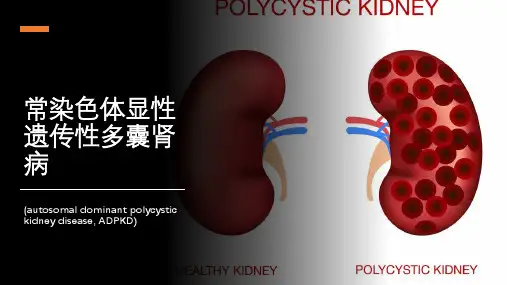
根据遗传方式不同, PKD可分为常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)和常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)。
ARPKD 多见于婴儿和儿童,发病率1/40000,多数早年夭折,很少存活至成年;ADPKD 多在成年后发病,发病率1/1000~1/400。
PKD有50%最终发展为终末期肾功能衰竭,约占终末期肾功能衰竭病因的10%,故近年来多囊肾病成为国际肾脏病领域研究的热点。
为早日攻克多囊肾病,国际上成立了多个协作组,如美国的多囊肾病研究基金会(PKR Foundation)[1]。
近年来也有许多研究表明该病可能为一种在易感人群中发生的感染性疾病。
随着分子生物学的发展,人类对多囊肾病有了一定的认识。
现综述如下。
一.先天性多囊肾分子遗传学基础目前已知的有3种基因突变导致了常染色体显性遗传多囊肾病,据报道其中2种最为常见,PKD1所占比例约85%;PKD2所占比例约15%;还有一种是PKD3,所占比率很小[2] 。


胡桃夹综合征(nutcracker phenomenon)即(左肾静脉压迫综合征),又称胡桃夹现象,好发于青春期至40岁左右的男性,儿童发病分布在4~7岁,多发年龄见于13~16岁。
人体的血管像四通八达的道路一样,是有一定走向的。
左肾静脉行走在腹主动脉和肠系膜上动脉之间,这两条动脉构成40~60度的夹角,左肾静脉刚好通过此夹角。
从解剖上看,右肾静脉径直注入下腔静脉,行程短而直。
而左肾静脉则需穿过腹主动脉和肠系膜上动脉之间的夹角,跨越腹主动脉前方始能注入下腔静脉,因此左肾静脉远较右肾静脉长。
正常时,肠系膜上动脉与腹主动脉之间的夹角被肠系膜、脂肪、淋巴结和腹膜等所充塞,使左肾静脉不致受到压挤。
当青春期发育较快、身高迅速增长、脊柱过度伸展、体形急剧变化或肾下垂等情况下,左肾静脉在这个夹角中的日子就不好过了,会受到挤压,引起血流变化和相应的临床症状。
症状:胡桃夹现象的主要症状是血尿和蛋白尿,其中无症状肉眼血尿更易发现。
血尿的原因是左肾静脉受压致肾静脉高压,左肾静脉扩张所引流的输尿管周围静脉与生殖静脉淤血、与肾集合系统发生异常交通,或部分静脉壁变薄破裂,引起非肾小球性血尿,还会发生睾丸静脉和卵巢静脉淤血而出现肋腹痛,并于立位或行走时加重。
另外男性还能发生精索静脉曲张。
此外有蛋白尿,不规则月经出血,高血压等。
此病的诊断标准为:一侧肾出血;尿红细胞形态为非肾小球性;尿中钙排泄量正常;膀胱镜检查为左侧输尿管口喷血或血性尿;腹部彩超或CT检查可见左肾静脉扩张等。
超声检查:超声对胡桃夹综合征的诊断有着明显的优势,超声检查时可清晰显示腹主动脉、肠系膜上动脉及左肾静脉的解剖情况,在不同横断面均可找到左肾静脉扩张近段的最大内径,测值准确,同时可观察并测量肠系膜上动脉与腹主动脉夹角变化。
彩超血流速度提供更准确的血流动力学变化,有助于此病诊断。
超声检查还能除外先天性畸形、外伤、肿瘤、结石、感染性疾病及血管异常等造成的血尿。
多囊肾多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。



遗传性肾病的基因检测及其治疗技术研究随着人类基因科技的迅猛发展,遗传性疾病的基因检测和治疗技术也日益成熟。
其中,遗传性肾病是一种常见的家族性疾病,对患者及家庭造成了巨大的负担,因此对其的基因检测及治疗技术研究显得尤为重要。
一、遗传性肾病的类型遗传性肾病按照基因突变的部位和影响,可以分为单基因遗传和多基因遗传两类。
单基因遗传包括最为常见的ADPKD(成人多囊肾病)、ARPKD(儿童多囊肾病)、Alport综合征等;而多基因遗传包括肾小管间质病、先天性肾小球性疾病等多种。
二、基因检测技术随着基因检测技术的逐渐成熟,已经可以通过对患者家族史及对基因的检测,来判断患者是否患有遗传性肾病。
其中,ADPKD 患者中,大约85%有PKD1基因的突变。
因此,对于成年的ADPKD患者,仅需检测PKD1基因,即可对其进行准确的判断;而对于18岁以下的患者,还需检测PKD2基因。
ARPKD患者则需要检测PKHD1基因的突变。
基因检测是一种无创、高效的诊断方法,可以让患者尽早接受到合适的治疗。
三、遗传性肾病的治疗技术目前,遗传性肾病的治疗技术主要包括保守治疗、手术治疗和药物治疗三种。
1. 保守治疗保守治疗主要是通过控制高血压、糖尿病等基础疾病,使肾脏不受进一步伤害。
但对于多数遗传性肾病患者而言,如果不采取其他治疗措施,肾脏功能会逐渐下降,甚至最终导致肾衰竭。
2. 手术治疗手术治疗主要包括硬膜外脑室分流术、肾移植等。
硬膜外脑室分流术是一种可以减轻颅内压力、改善脑功能的手术,适用于脑积水等疾病。
而肾移植则是目前治疗肾衰竭的常规治疗方法,对于遗传性肾病患者,肾移植可以起到延缓肾脏损害的作用。
3. 药物治疗药物治疗主要是通过调节肾脏的生理功能,延缓肾脏的进一步损害。
而目前,新一代的基因治疗技术也已经开始应用于临床,对于ADPKD等囊肿性肾病患者,可以通过小分子化合物等药物来针对性地抑制囊肿增长。
四、结语遗传性肾病是一种严重的疾病,对患者、家庭和社会造成了极大的负担。


多囊肾多囊肾病(polycystic kidney disease,PKD)是一种常见的遗传性肾脏病,主要表现为双侧肾脏出现多个大小不一的囊肿,囊肿进行性增大,最终破坏肾脏结构和功能,导致终末期肾功能衰竭。
根据遗传方式不同,可分为常染色体显性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)和常染色隐性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)。
常染色体隐性遗传型多囊肾,发病于婴儿期,临床较罕见;常染色体显性遗传型多囊肾,常于青中年时期被发现,也可在任何年龄发病。
ADPKD是一种最常见的单基因遗传性肾病,发病率1/1000~1/4000,发病年龄多在30~50岁,故既往又称之为“成人型多囊肾病”,实际上该病可发生于任何年龄,甚至胎儿,故“成人型”这一术语并不准确,现已废用。
ADPKD除累及肾脏外,还可伴有肝囊肿、胰腺囊肿、颅内动脉瘤、心脏瓣膜异常等,因此,它也是一种系统性疾病。
目前已经明确引起多囊肾病的突变基因主要有PKD1HE PKD2两种。
60岁以上患者将有50%将发展至终末期肾衰竭,占终末期肾衰竭病因的5~10%。
ARPKD是一种隐性遗传性肾病,一般在婴儿期即有明显表现,因此过去称为“婴儿型多囊肾病”,少部分发生于儿童或青少年。
发病率约1/1万~1/4万,常伴有肝脏受累,表现为肝囊肿。
目前已发现其发病与PKHD1基因有关。
ARPKD患儿中,50%在出生后数小时至数天内死于呼吸衰竭或肾衰竭,存活至成人者主要特征是肾集合管纺锤形扩张,进展至肾衰竭,同时伴有肝内胆管扩张、先天性肝纤维化,临床表现为门脉高压症。
由于ARPKD是一种少见病,多发生于儿童,故本文仅介绍ADPKD。
本病为常染色体显性遗传,按其遗传规律,代代发病,男女患病几率均等。
父母一方患病,子女发病几率50%。

多囊肾演示课件xx年xx月xx日CATALOGUE目录•简介•临床表现•诊断与鉴别诊断•治疗•预后及预防•研究进展及展望01简介多囊肾是一种常见的遗传性肾脏疾病,由肾小管上皮细胞异常增生和聚集引起,以双侧肾脏多个囊肿形成为主要特征。
定义多数多囊肾病例为基因突变导致,其中约1/3为新发突变,2/3为家族遗传。
病因什么是多囊肾婴儿型多囊肾常染色体隐性遗传,多在出生后不久确诊,病变严重发展较快,易出现肾功能不全。
成人型多囊肾常染色体显性遗传,多为成年后发病,病变进展速度差异较大,有的患者病变轻微,肾功能正常。
多囊肾的分类1多囊肾的遗传方式23成人型多囊肾常为常染色体显性遗传,患者的一级亲属中约有一半可能患病。
常染色体显性遗传婴儿型多囊肾多为常染色体隐性遗传,患者父母均正常,但兄弟姐妹中可能有患者。
常染色体隐性遗传多囊肾的遗传方式不同,家族中其他成员患病情况也不同,但均为垂直遗传。
遗传特点02临床表现成人型多囊肾患者肾脏明显肿大,可压迫周围器官,引起腰部不适、疼痛、呼吸困难等症状。
肾脏肿大随着病情进展,患者肾功能逐渐减退,出现尿毒症、高血压、肾衰竭等严重并发症。
肾功能减退患者还可能出现贫血、骨质疏松、矿物质代谢紊乱等症状。
其他症状婴儿型多囊肾患者肾脏在胎儿期就出现肿大,可压迫胎儿胃肠道,引起胃部不适、恶心、呕吐等症状。
肾脏肿大婴儿型多囊肾患者肾功能衰竭进展迅速,出生后不久即出现严重肾功能不全,最终可导致死亡。
肾功能衰竭患者还可能出现羊水过多、呼吸困难、低血糖等症状。
其他症状03其他症状患者还可能出现眼部异常、颅面部畸形、心血管疾病、神经系统疾病等多种症状。
基因突变致多囊肾疾病的临床表现01遗传方式多样化基因突变致多囊肾疾病遗传方式多种多样,包括常染色体显性遗传、常染色体隐性遗传、X连锁隐性遗传等。
02肾功能受损患者肾功能受损程度差异较大,有的患者肾功能正常,有的则出现严重肾功能不全。
03诊断与鉴别诊断临床表现多囊肾的成年患者通常在40岁左右出现症状,表现为疼痛、腹部肿块和肾功能损害等。

*基金项目:江西省卫生计生委科技计划项目(20161045)①南昌大学第二附属医院 江西 南昌 330006通信作者:陈艳染色体显性遗传性多囊肾病PKD2基因突变的检测*陈艳① 黄翀① 徐承云① 【摘要】 目的:建立检测常染色体显性遗传性多囊肾病2型(polycystic kidney disease 2,PKD2)致病基因突变的方法,检测收集的10个ADPKD 家系共15例患者的PKD2基因突变情况。
方法:收集江西地区经临床确诊的常染色体显性遗传性多囊肾病患者15例,采集外周静脉血5 mL,用试剂盒提取基因组DNA,采用聚合酶链式反应(PCR)扩增PKD2基因全部外显子及相近内含子区域,PCR 扩增产物经分离纯化后直接进行基因序列测序,根据测序图谱进行突变分析,明确基因突变位点和类型。
结果:15例常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)患者中,检测出3个正常基因多态性位点,分别为1号外显子第420位碱基G 置换为A,未引起编码氨基酸改变;1号外显子第568位碱基G 置换为A,致使190位编码氨基酸由丙氨酸改变为苏氨酸;内含子靠近cDNA 第844位碱基5’端的第22个碱基A 置换为G。
结论:可通过直接基因序列测定对PKD2进行基因突变检测,本研究所检测到的3个正常基因多态性位点均已有报道,为开展ADPKD 直接基因诊断、产前诊断及症状前诊断提供了实验基础。
【关键词】 常染色体显性遗传性多囊肾病; PKD2; 基因突变; 多态性 Detection of PKD2 Gene Mutation in Chromosomal Dominant Polycystic Kidney Disease/CHEN Yan,HUANG Chong,XU Chengyun.//Medical Innovation of China,2019,16(26):145-150 【Abstract】 Objective:To set up a method for detecting the mutations of autosomal dominant olycystic kidney disease gene 2, detect the mutations of PKD2 in 15 patients from 10 ADPKD families.Method:Fifteen patients with autosomal dominant polycystic kidney disease diagnosed clinically in Jiangxi were collected,5 mL peripheral venous blood was collected and genomic DNA was extracted with the kit.All exons and close intron regions of PKD2 gene were amplified by polymerase chain reaction(PCR),after isolation and purification,PCR amplification products were directly sequenced,and mutation analysis was conducted according to the sequencing map to identify the mutation sites and types of genes.Result:Three polymorphisms were detected in 15 ADPKD patients.The c.568G>A in exon1 caused the translation change(Ala190Thr).The other two (c.420G>A in exon1 and 844-22A>G in IVS3) did not cause the translation change.Conclusion:Gene mutation detection of PKD2 can be performed by direct gene sequencing,all the 3 normal gene polymorphisms detected in this study have been reported, providing an experimental basis for direct gene diagnosis, prenatal diagnosis and pre-symptom diagnosis of ADPKD. 【Key words】 Autosomal dominant polycystic kidney disease; PKD2; Gene mutation; Polymorphism First-author ’s address:Second Affiliated Hospital of Nanchang University,Nanchang 330006,China doi:10.3969/j.issn.1674-4985.2019.26.039 常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)又称为成人型多囊肾病,是最常见的肾脏遗传病,发病率为1/400~1/1 000[1],我国约有150万例患者。