11食品中元素含量的测定
- 格式:ppt
- 大小:480.50 KB
- 文档页数:50

实验八:食品中铅、镉、铬的测定(原子吸收光谱法综合性试验)一、目的与要求1.通过实际试样,对食品中的多种限量金属成分,采用不同的光谱分析条件进行测定,以达到综合应用原子吸收光谱法的目的。
2.根据各元素的分析特性,试样的含量,基体组成及可能干扰选取合适的分析条件。
包括了试样的制备、预处理、标准溶液的配制及校正曲线的制作、分析条件的选择、操作方法、结果计算、数据处理及误差分析等。
二、实验原理与相关知识食品中有害金属元素铅、镉、铬的测定,目前国际上通用的方法均以石墨炉原子化法较为准确、快速。
该法检出限为5μg/kg,基于基态自由原子对特定波长光吸收的一种测量方法,它的基本原理是使光源辐射出的待测元素的特征光谱通过样品的蒸汽时,被蒸汽中待测元素的基态原子所吸收,在一定范围与条件下,入射光被吸收而减弱的程度与样品中待测元素的含量成正比关系,由此可得出样品中待测元素的含量。
食品中铅、镉、铬等元素的基态原子对空心阴极灯的共辐射都有选择性吸收,但是各元素具体的分析条件不同,例如铅的测定是氧化性气氛,但铬的测定却要求还原性气氛,并且要有高性能的空心阴极灯才能获得足够的灵敏度。
这些元素的灵敏度都有差别,因此配制标准序列时,浓度序列有所不同,但是它们在一定的浓度下,彼此不会干扰,因而可以把它们的标准溶液混合配在一起,方便操作。
三、仪器与试剂1.实验室提供的仪器与试剂(1)石墨炉原子吸收分光光度计(具氘灯扣背景装置)及其它配件;(2)氮气钢瓶;(3)铅、镉、铬等元素空心阴极灯。
(4)基准试剂:铅、镉、铬标准贮备液。
(5)基体改进试剂:①磷酸二氨铵溶液(20g/L);②盐酸溶液(1mol/L);③柠檬酸钠缓冲溶液(2mol/L);④双硫腙-乙酸丁酯溶液(0.1%);⑤二乙基二硫代氨基甲酸钠溶液(1%)2.由学生自配试剂(1) 铅、镉、铬系列标准使用溶液;(2) 样品消化及定容用试剂。
四、实验方案的设计提示1.测定各元素离子时样品的处理方案样品经消解(可选用干法灰化、压力消解法、常压湿法消化、微波消解法中的任何一种)后,制成供试样液,可参考表5-3-12.测定各元素离子的标准系列配制方案:可参考表表5-3-2或参考其他资料.3.测定铅、镉、铬的条件选择:由于仪器型号规格不同,测定条件有所差别,可根据仪器说明书选择最佳条件测试,可参考表5-3-3. 表5-3-1 食品中铅、镉、铬测定用样品的处理表5-3-2 金属离子标准溶液系列五、实验步骤(按设计的方案进行实验)(一) 样品处理(可参照表5-3-1 食品中铅、镉、铬测定用样品的处理) (二) 操作步骤参照仪器说明书,根据各自仪器性能及设计的方案调至最佳状态,简要步骤如下: 1. 安装待测元素空心阴极灯,对准位置,固定待测波长及狭缝宽度。



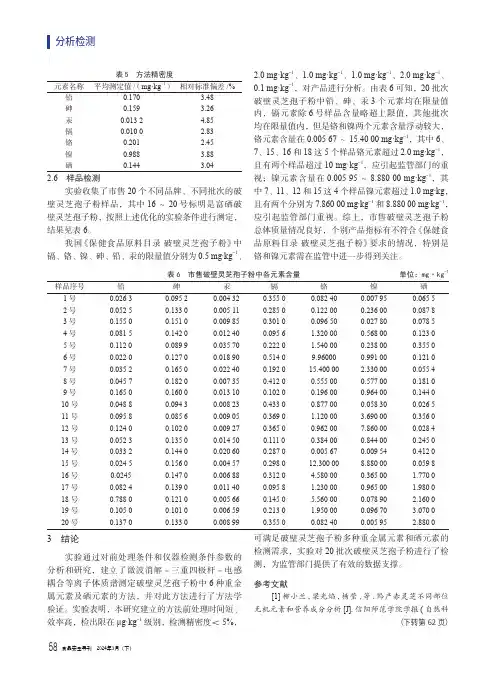
表5 方法精密度元素名称平均测定值/(mg·kg-1)相对标准偏差/%铅0.170 3.48砷0.159 3.26汞0.013 2 4.85镉0.010 0 2.83铬0.201 2.45镍0.988 3.88硒0.144 3.042.6 样品检测实验收集了市售20个不同品牌、不同批次的破壁灵芝孢子粉样品,其中16~20号标明是富硒破壁灵芝孢子粉,按照上述优化的实验条件进行测定,结果见表6。
我国《保健食品原料目录破壁灵芝孢子粉》中镉、铬、镍、砷、铅、汞的限量值分别为0.5 mg·kg-1、2.0 mg·kg-1、1.0 mg·kg-1、1.0 mg·kg-1、2.0 mg·kg-1、0.1 mg·kg-1,对产品进行分析。
由表6可知,20批次破壁灵芝孢子粉中铅、砷、汞3个元素均在限量值内,镉元素除6号样品含量略超上限值,其他批次均在限量值内,但是铬和镍两个元素含量浮动较大,铬元素含量在0.005 67~15.40 00 mg·kg-1,其中6、7、15、16和18这5个样品铬元素超过2.0 mg·kg-1,且有两个样品超过10 mg·kg-1,应引起监管部门的重视;镍元素含量在0.005 95~8.880 00 mg·kg-1,其中7、11、12和15这4个样品镍元素超过1.0 mg·kg,且有两个分别为7.860 00 mg·kg-1和8.880 00 mg·kg-1,应引起监管部门重视。
综上,市售破壁灵芝孢子粉总体质量情况良好,个别产品指标有不符合《保健食品原料目录破壁灵芝孢子粉》要求的情况,特别是铬和镍元素需在监管中进一步得到关注。
表6 市售破壁灵芝孢子粉中各元素含量 单位:mg·kg-1样品序号铅砷汞镉铬镍硒1号0.026 30.095 20.004 320.355 00.082 400.007 950.065 5 2号0.052 50.133 00.005 110.285 00.122 000.236 000.087 8 3号0.155 00.151 00.009 850.301 00.096 500.027 800.078 5 4号0.081 50.142 00.012 400.095 6 1.320 000.568 000.123 0 5号0.112 00.089 90.035 700.222 0 1.540 000.238 000.355 0 6号0.022 00.127 00.018 900.514 09.960000.991 000.121 0 7号0.035 20.165 00.022 400.192 015.400 00 2.330 000.055 4 8号0.045 70.182 00.007 350.412 00.555 000.577 000.181 0 9号0.165 00.160 00.013 100.102 00.196 000.964 000.144 0 10号0.048 80.094 30.008 230.433 00.877 000.058 300.026 5 11号0.095 80.085 60.009 050.369 0 1.120 00 3.690 000.356 0 12号0.124 00.102 00.009 270.365 00.962 007.860 000.028 4 13号0.052 30.135 00.014 500.111 00.384 000.844 000.245 0 14号0.033 20.144 00.020 600.287 00.005 670.009 540.412 0 15号0.024 50.156 00.004 570.298 012.300 008.880 000.059 8 16号0.02450.147 00.006 880.312 0 4.580 000.365 00 1.770 0 17号0.082 40.139 00.011 400.095 8 1.230 000.965 00 1.980 0 18号0.788 00.121 00.005 660.145 0 5.560 000.078 90 2.160 0 19号0.105 00.101 00.006 590.213 0 1.950 000.096 70 3.070 0 20号0.137 00.133 00.008 990.355 00.082 400.005 95 2.880 03 结论实验通过对前处理条件和仪器检测条件参数的分析和研究,建立了微波消解-三重四极杆-电感耦合等离子体质谱测定破壁灵芝孢子粉中6种重金属元素及硒元素的方法,并对此方法进行了方法学验证。

食品中微量兀素的常规检验方法食品中微量兀素的常规检验方法摘要:现如今人们对食品安全问题越来越重视,对社会报道的食品安全事件较为关注,尤其是对于食品中微量元素的污染问题,逐渐成为人类健康的核心影响因素之一,对食品安全有严重的威胁。
因此,食品中微量元素的测定已成为当前食品安全检查中的核心工作内容。
但我国与发达国家的食品安全测定与问题分析相比较而言还存在较大的差距。
关键词:食品安全微量元素检验测定引言:随着国民经济的快速发展,食品安全问题已经成为我国发展过程中需要面临的重要难题和挑战,对于政府的食品安全检测部门和生产企业都是一个巨大的考验。
我国现有的食品中微量元素的检测方式已经不能满足现代社会发展的需要,迫切需要完善的检验方式,一门新兴的边缘化科学“生命科学中的微量元素”由此应运而生。
本文就当前常规的食品微量元素的检验方法及其测定的重要性进行分析探讨。
一、原子荧光光谱法每种元素的原子荧光强度都是特定的,根据此原理就可以检验出待测的元素含量。
这种方法的特点是检测的灵敏度较高,实施过程中的干扰比较少,具有较宽的线性范围,并且能够将较多的元素放在一起同时检测分析。
NaBH4与汞离子、SnCL2与汞离子都可以反应形成原子态的汞,在室温环境中能够被相互作用从而变成汞原子荧光,这种方式叫做冷原子荧光光谱法,也可以称作冷蒸汽法。
因为AFS的测定方法对检验汞的敏感程度较高,所以在分析样品汞含量的时候通常较多的运用冷原子荧光与无焰、有焰HG-AFG^几种测定方式。
如果想要检验大米当中的汞元素就可以使用原子荧光光谱法,它是通过微波加热的方式使样品在温度较高和压力较大的环境下消解样品。
同时也可以利用此方法检验错这一微量元素,它多存在于保健食品当中。
可以研究酸介质和氢氧化物等因素对检验所产生的影响。
把仪器最适宜的工作条件选定出来,用酒石酸来进行检验,不仅能够打破干扰因素共存离子,还能够增敏,可以适用于在室温原子化环境下检验错在保健食品中的痕量。

食品中重金属残留的检测方法随着人们对健康的重视程度逐渐提高,食品安全问题成为了人们关注的重点之一。
其中,食品中的重金属残留问题备受关注。
重金属是指比较密度较大,具有较强的毒性和生物积累性的元素,如铅、汞、镉、砷等。
食品中的重金属残留会对人体造成很大的危害,因此必须对食品中的重金属含量进行检测。
下面将介绍一些常见的食品中重金属残留的检测方法。
一、原子吸收光谱法(AAS)原子吸收光谱法是一种分析化学方法,可用于检测食品中的重金属残留。
该方法的基本原理是利用原子对吸收较明显的某种波长的光的量与元素浓度之间的关系来分析元素。
AAS法具有灵敏度高、专属性强、分析时间短、误差小等优点,但是该法的适用性和灵敏度仅限于特定元素,且样品处理方法较为繁琐。
二、电感耦合等离子体发射光谱法(ICP-OES)电感耦合等离子体发射光谱法是一种分析化学方法,用于分析食品中的重金属元素含量。
该方法的原理是利用样品在高温下气化,生成几千度高温下的等离子体,再用光电多道辐射计测定不同波长的辐射强度,进而分析样品中重金属元素的含量。
ICP-OES 法具有分析速度快、灵敏度高、准确度好等优点,但是设备较为昂贵,需要专业技术人员操作。
三、电感耦合等离子体质谱法(ICP-MS)电感耦合等离子体质谱法是一种高灵敏度的分析方法,可用于检测食品中的重金属元素含量。
该方法的原理是利用设备将样品中元素离子化成带正电荷的离子,并测定离子的质量和相对丰度,进而分析样品中重金属元素的含量。
ICP-MS法具有极高的灵敏度和准确度,但是设备价格昂贵,需要专业技术人员操作。
四、原子荧光光谱法(AFS)原子荧光光谱法是一种基于元素的原子荧光现象实现的分析技术,可用于检测食品中的重金属元素含量。
该方法的基本原理是通过激发样品中重金属元素的原子产生荧光,然后测定荧光的强度,从而确定元素的含量。
AFS法具有高准确性、精度高、测定速度快等特点,但是分析的元素种类相对较少,且需要较为严格的样品预处理。

第1篇一、实验目的本次实验旨在通过化学分析和仪器分析方法,检测食品中的主要成分,包括蛋白质、脂肪、碳水化合物、矿物质、维生素等,以了解食品的营养成分和卫生状况。
二、实验原理1. 蛋白质:采用双缩脲法测定蛋白质含量,原理为蛋白质中的肽键与双缩脲试剂发生反应,生成紫色络合物,其颜色深浅与蛋白质含量成正比。
2. 脂肪:采用索氏抽提法测定脂肪含量,原理为脂肪不溶于水,可被有机溶剂抽提,通过称量抽提出的脂肪量来计算脂肪含量。
3. 碳水化合物:采用苯酚-硫酸法测定碳水化合物含量,原理为碳水化合物与苯酚在硫酸作用下生成蓝绿色化合物,其颜色深浅与碳水化合物含量成正比。
4. 矿物质:采用原子吸收光谱法测定矿物质含量,原理为样品中的矿物质元素在特定波长下吸收特定波长的光,通过测定吸光度来计算元素含量。
5. 维生素:采用高效液相色谱法测定维生素含量,原理为维生素具有特定的紫外吸收峰,通过测定样品的紫外吸收光谱来计算维生素含量。
三、实验材料1. 样品:面粉、牛奶、鸡蛋、水果等。
2. 试剂:双缩脲试剂、苯酚、硫酸、盐酸、氢氧化钠、三氯化铁、氯化钠、硝酸、高氯酸、原子吸收光谱标准溶液、维生素标准溶液等。
3. 仪器:分光光度计、索氏抽提器、电子天平、原子吸收光谱仪、高效液相色谱仪等。
四、实验步骤1. 蛋白质含量测定(1)称取样品0.5g,加入5mL双缩脲试剂,充分混合,室温放置10min。
(2)以试剂空白为参比,在540nm波长下测定吸光度。
(3)根据标准曲线计算蛋白质含量。
2. 脂肪含量测定(1)称取样品5g,加入50mL石油醚,在索氏抽提器中抽提6h。
(2)将抽提液转移至锥形瓶中,挥去石油醚,加入5mL盐酸,加热至近干。
(3)加入5mL氢氧化钠溶液,充分混合,加入5mL三氯化铁溶液,静置10min。
(4)以试剂空白为参比,在520nm波长下测定吸光度。
(5)根据标准曲线计算脂肪含量。
3. 碳水化合物含量测定(1)称取样品5g,加入10mL苯酚溶液,充分混合。

食品安全国家标准食品中钙的测定1范围本标准规定了食品中钙含量测定的火焰原子吸收光谱法㊁滴定法㊁电感耦合等离子体发射光谱法和电感耦合等离子体质谱法㊂本标准适用于食品中钙含量的测定㊂第一法火焰原子吸收光谱法2原理试样经消解处理后,加入镧溶液作为释放剂,经原子吸收火焰原子化,在422.7n m处测定的吸光度值在一定浓度范围内与钙含量成正比,与标准系列比较定量㊂3试剂和材料除非另有规定,本方法所用试剂均为优级纯,水为G B/T6682规定的二级水㊂3.1试剂3.1.1硝酸(H N O3)㊂3.1.2高氯酸(H C l O4)㊂3.1.3盐酸(H C l)㊂3.1.4氧化镧(L a2O3)㊂3.2试剂配制3.2.1硝酸溶液(5+95):量取50m L硝酸,加入950m L水,混匀㊂3.2.2硝酸溶液(1+1):量取500m L硝酸,与500m L水混合均匀㊂3.2.3盐酸溶液(1+1):量取500m L盐酸,与500m L水混合均匀㊂3.2.4镧溶液(20g/L):称取23.45g氧化镧,先用少量水湿润后再加入75m L盐酸溶液(1+1)溶解,转入1000m L容量瓶中,加水定容至刻度,混匀㊂3.3标准品碳酸钙(C a C O3,C A S号471-34-1):纯度>99.99%,或经国家认证并授予标准物质证书的一定浓度的钙标准溶液㊂3.4标准溶液的配制3.4.1钙标准储备液(1000m g/L):准确称取2.4963g(精确至0.0001g)碳酸钙,加盐酸溶液(1+1)溶解,移入1000m L容量瓶中,加水定容至刻度,混匀㊂3.4.2钙标准中间液(100m g/L):准确吸取钙标准储备液(1000m g/L)10m L于100m L容量瓶中,加硝酸溶液(5+95)至刻度,混匀㊂3.4.3钙标准系列溶液:分别吸取钙标准中间液(100m g/L)0m L,0.500m L,1.00m L,2.00m L,4.00m L,6.00m L于100m L容量瓶中,另在各容量瓶中加入5m L镧溶液(20g/L),最后加硝酸溶液(5+95)定容至刻度,混匀㊂此钙标准系列溶液中钙的质量浓度分别为0m g/L㊁0.500m g/L㊁1.00m g/L㊁2.00m g/L㊁4.00m g/L和6.00m g/L㊂注:可根据仪器的灵敏度及样品中钙的实际含量确定标准溶液系列中元素的具体浓度㊂4仪器设备注:所有玻璃器皿及聚四氟乙烯消解内罐均需硝酸溶液(1+5)浸泡过夜,用自来水反复冲洗,最后用水冲洗干净㊂4.1原子吸收光谱仪:配火焰原子化器,钙空心阴极灯㊂4.2分析天平:感量为1m g和0.1m g㊂4.3微波消解系统:配聚四氟乙烯消解内罐㊂4.4可调式电热炉㊂4.5可调式电热板㊂4.6压力消解罐:配聚四氟乙烯消解内罐㊂4.7恒温干燥箱㊂4.8马弗炉㊂5分析步骤5.1试样制备注:在采样和试样制备过程中,应避免试样污染㊂5.1.1粮食㊁豆类样品样品去除杂物后,粉碎,储于塑料瓶中㊂5.1.2蔬菜㊁水果㊁鱼类㊁肉类等样品样品用水洗净,晾干,取可食部分,制成匀浆,储于塑料瓶中㊂5.1.3饮料㊁酒㊁醋㊁酱油㊁食用植物油㊁液态乳等液体样品将样品摇匀㊂5.2试样消解5.2.1湿法消解准确称取固体试样0.2g~3g(精确至0.001g)或准确移取液体试样0.500m L~5.00m L于带刻度消化管中,加入10m L硝酸㊁0.5m L高氯酸,在可调式电热炉上消解(参考条件:120ħ/0.5h~120ħ/1h㊁升至180ħ/2h~180ħ/4h㊁升至200ħ~220ħ)㊂若消化液呈棕褐色,再加硝酸,消解至冒白烟,消化液呈无色透明或略带黄色㊂取出消化管,冷却后用水定容至25m L,再根据实际测定需要稀释,并在稀释液中加入一定体积的镧溶液(20g/L),使其在最终稀释液中的浓度为1g/L,混匀备用,此为试样待测液㊂同时做试剂空白试验㊂亦可采用锥形瓶,于可调式电热板上,按上述操作方法进行湿法消解㊂5.2.2微波消解准确称取固体试样0.2g~0.8g(精确至0.001g)或准确移取液体试样0.500m L~3.00m L于微波消解罐中,加入5m L硝酸,按照微波消解的操作步骤消解试样,消解条件参考附录A㊂冷却后取出消解罐,在电热板上于140ħ~160ħ赶酸至1m L左右㊂消解罐放冷后,将消化液转移至25m L容量瓶中,用少量水洗涤消解罐2次~3次,合并洗涤液于容量瓶中并用水定容至刻度㊂根据实际测定需要稀释,并在稀释液中加入一定体积镧溶液(20g/L)使其在最终稀释液中的浓度为1g/L,混匀备用,此为试样待测液㊂同时做试剂空白试验㊂5.2.3压力罐消解准确称取固体试样0.2g~1g(精确至0.001g)或准确移取液体试样0.500m L~5.00m L于消解内罐中,加入5m L硝酸㊂盖好内盖,旋紧不锈钢外套,放入恒温干燥箱,于140ħ~160ħ下保持4h~ 5h㊂冷却后缓慢旋松外罐,取出消解内罐,放在可调式电热板上于140ħ~160ħ赶酸至1m L左右㊂冷却后将消化液转移至25m L容量瓶中,用少量水洗涤内罐和内盖2次~3次,合并洗涤液于容量瓶中并用水定容至刻度,混匀备用㊂根据实际测定需要稀释,并在稀释液中加入一定体积的镧溶液(20g/L),使其在最终稀释液中的浓度为1g/L,混匀备用,此为试样待测液㊂同时做试剂空白试验㊂5.2.4干法灰化准确称取固体试样0.5g~5g(精确至0.001g)或准确移取液体试样0.500m L~10.0m L于坩埚中,小火加热,炭化至无烟,转移至马弗炉中,于550ħ灰化3h~4h㊂冷却,取出㊂对于灰化不彻底的试样,加数滴硝酸,小火加热,小心蒸干,再转入550ħ马弗炉中,继续灰化1h~2h,至试样呈白灰状,冷却,取出,用适量硝酸溶液(1+1)溶解转移至刻度管中,用水定容至25m L㊂根据实际测定需要稀释,并在稀释液中加入一定体积的镧溶液,使其在最终稀释液中的浓度为1g/L,混匀备用,此为试样待测液㊂同时做试剂空白试验㊂5.3仪器参考条件参考条件见附录B㊂5.4标准曲线的制作将钙标准系列溶液按浓度由低到高的顺序分别导入火焰原子化器,测定吸光度值,以标准系列溶液中钙的质量浓度为横坐标,相应的吸光度值为纵坐标,制作标准曲线㊂5.5试样溶液的测定在与测定标准溶液相同的实验条件下,将空白溶液和试样待测液分别导入原子化器,测定相应的吸光度值,与标准系列比较定量㊂6分析结果的表述试样中钙的含量按式(1)计算:X=(ρ-ρ0)ˑfˑVm(1)式中:X 试样中钙的含量,单位为毫克每千克或毫克每升(m g/k g或m g/L);ρ 试样待测液中钙的质量浓度,单位为毫克每升(m g/L);ρ0 空白溶液中钙的质量浓度,单位为毫克每升(m g/L);f 试样消化液的稀释倍数;V 试样消化液的定容体积,单位为毫升(m L);m 试样质量或移取体积,单位为克或毫升(g或m L)㊂当钙含量ȡ10.0m g/k g或10.0m g/L时,计算结果保留三位有效数字,当钙含量<10.0m g/k g或10.0m g/L时,计算结果保留两位有效数字㊂7精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%㊂8其他以称样量0.5g(或0.5m L),定容至25m L计算,方法检出限为0.5m g/k g(或0.5m g/L),定量限为1.5m g/k g(或1.5m g/L)㊂第二法E D T A滴定法9原理在适当的p H范围内,钙与E D T A(乙二胺四乙酸二钠)形成金属络合物㊂以E D T A滴定,在达到当量点时,溶液呈现游离指示剂的颜色㊂根据E D T A用量,计算钙的含量㊂10试剂和材料除非另有规定,本方法所用试剂均为分析纯,水为G B/T6682规定的三级水㊂10.1试剂10.1.1氢氧化钾(K O H)㊂10.1.2硫化钠(N a2S)㊂10.1.3柠檬酸钠(N a3C6H5O7㊃2H2O)㊂10.1.4乙二胺四乙酸二钠(E D T A,C10H14N2O8N a2㊃2H2O)㊂10.1.5盐酸(H C l):优级纯㊂10.1.6钙红指示剂(C21O7N2S H14)㊂10.1.7硝酸(H N O3):优级纯㊂10.1.8高氯酸(H C l O4):优级纯㊂10.2试剂配制10.2.1氢氧化钾溶液(1.25m o l/L):称取70.13g氢氧化钾,用水稀释至1000m L,混匀㊂10.2.2硫化钠溶液(10g/L):称取1g硫化钠,用水稀释至100m L,混匀㊂10.2.3柠檬酸钠溶液(0.05m o l/L):称取14.7g柠檬酸钠,用水稀释至1000m L,混匀㊂10.2.4 E D T A溶液:称取4.5g E D T A,用水稀释至1000m L,混匀,贮存于聚乙烯瓶中,4ħ保存㊂使用时稀释10倍即可㊂10.2.5钙红指示剂:称取0.1g钙红指示剂,用水稀释至100m L,混匀㊂10.2.6盐酸溶液(1+1):量取500m L盐酸,与500m L水混合均匀㊂10.3标准品碳酸钙(C a C O3,C A S号471-34-1):纯度>99.99%,或经国家认证并授予标准物质证书的一定浓度的钙标准溶液㊂10.4标准溶液配制钙标准储备液(100.0m g/L):准确称取0.2496g(精确至0.0001g)碳酸钙,加盐酸溶液(1+1)溶解,移入1000m L容量瓶中,加水定容至刻度,混匀㊂11仪器设备注:所有玻璃器皿均需硝酸溶液(1+5)浸泡过夜,用自来水反复冲洗,最后用水冲洗干净㊂11.1分析天平:感量为1m g和0.1m g㊂11.2可调式电热炉㊂11.3可调式电热板㊂11.4马弗炉㊂12分析步骤12.1试样制备同5.1㊂12.2试样消解12.2.1湿法消解同5.2.1㊂12.2.2干法灰化同5.2.4㊂12.3滴定度(T)的测定吸取0.500m L钙标准储备液(100.0m g/L)于试管中,加1滴硫化钠溶液(10g/L)和0.1m L柠檬酸钠溶液(0.05m o l/L),加1.5m L氢氧化钾溶液(1.25m o l/L),加3滴钙红指示剂,立即以稀释10倍的E D T A溶液滴定,至指示剂由紫红色变蓝色为止,记录所消耗的稀释10倍的E D T A溶液的体积㊂根据滴定结果计算出每毫升稀释10倍的E D T A溶液相当于钙的毫克数,即滴定度(T)㊂12.4试样及空白滴定分别吸取0.100m L~1.00m L(根据钙的含量而定)试样消化液及空白液于试管中,加1滴硫化钠溶液(10g/L)和0.1m L柠檬酸钠溶液(0.05m o l/L),加1.5m L氢氧化钾溶液(1.25m o l/L),加3滴钙红指示剂,立即以稀释10倍的E D T A溶液滴定,至指示剂由紫红色变蓝色为止,记录所消耗的稀释10倍的E D T A溶液的体积㊂13分析结果的表述试样中钙的含量按式(2)计算:X=Tˑ(V1-V0)ˑV2ˑ1000mˑV3(2)式中:X 试样中钙的含量,单位为毫克每千克或毫克每升(m g/k g或m g/L);T E D T A滴定度,单位为毫克每毫升(m g/m L);V1 滴定试样溶液时所消耗的稀释10倍的E D T A溶液的体积,单位为毫升(m L);V0 滴定空白溶液时所消耗的稀释10倍的E D T A溶液的体积,单位为毫升(m L);V2 试样消化液的定容体积,单位为毫升(m L);1000 换算系数;m 试样质量或移取体积,单位为克或毫升(g或m L);V3 滴定用试样待测液的体积,单位为毫升(m L)㊂计算结果保留三位有效数字㊂14精密度在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%㊂15其他以称样量4g(或4m L),定容至25m L,吸取1.00m L试样消化液测定时,方法的定量限为100m g/k g(或100m g/L)㊂第三法电感耦合等离子体发射光谱法见G B5009.268㊂第四法电感耦合等离子体质谱法见G B5009.268㊂附录A微波消解升温程序参考条件微波消解升温程序参考条件见表A.1㊂表A.1微波消解升温程序参考条件步骤设定温度ħ升温时间m i n恒温时间m i n112055 2160510 3180510附录B火焰原子吸收光谱法参考条件火焰原子吸收光谱法参考条件见表B.1㊂表B.1火焰原子吸收光谱法参考条件元素波长n m狭缝n m灯电流m A燃烧头高度mm空气流量L/m i n乙炔流量L/m i n钙422.71.35~15392。

食品中铅的测定:第一法石墨炉原子吸收光谱法3 原理试样经灰化或酸消解后,注入原子吸收分光光度计石墨炉中,电热原子化后吸收283。
3 nm 共振线,在一定浓度范围,其吸收值与铅含量成正比,与标准系列比较定量。
4 试剂和材料硝酸:优级纯。
4.2 过硫酸铵。
4。
3 过氧化氢(30%)。
4。
4 高氯酸:优级纯。
4.5 硝酸(1+1):取50 mL 硝酸慢慢加入50 mL 水中。
4。
6 硝酸(0.5 mol/L):取3.2 mL 硝酸加入50 mL 水中,稀释至100 mL。
4。
7 硝酸(l mo1/L):取6.4 mL 硝酸加入50 mL 水中,稀释至100 mL。
4。
8 磷酸二氢铵溶液(20 g/L):称取2。
0 g 磷酸二氢铵,以水溶解稀释至100 mL。
4。
9 混合酸:硝酸十高氯酸(9+1)。
取9 份硝酸与1 份高氯酸混合。
4.10 铅标准储备液:准确称取1。
000 g 金属铅(99.99%),分次加少量硝酸(4。
5),加热溶解,总量不超过37 mL,移入1000 mL 容量瓶,加水至刻度。
混匀。
此溶液每毫升含 1.0 mg 铅.4。
11 铅标准使用液:每次吸取铅标准储备液1。
0 mL 于100 mL 容量瓶中,加硝酸(4.6)至刻度。
如此经多次稀释成每毫升含10。
0 ng,20.0 ng,40。
0 ng,60。
0 ng,80.0 ng 铅的标准使用液。
5 仪器和设备5。
1 原子吸收光谱仪,附石墨炉及铅空心阴极灯。
5。
2 马弗炉。
5。
3 天平:感量为1 mg.5。
4 干燥恒温箱.5。
5 瓷坩埚。
5.6 压力消解器、压力消解罐或压力溶弹。
5.7 可调式电热板、可调式电炉。
6 分析步骤6.2 试样消解(可根据实验室条件选用以下任何一种方法消解)6.2.1 湿式消解法:称取试样1 g~5 g(精确到0.001 g)于锥形瓶或高脚烧杯中,放数粒玻璃珠,加10 mL 混合酸(4。
9),加盖浸泡过夜,加一小漏斗于电炉上消解,若变棕黑色,再加混合酸,直至冒白烟,消化液呈无色透明或略带黄色,放冷,用滴管将试样消化液洗入或过滤入(视消化后试样的盐分而定)10 mL~25 mL 容量瓶中,用水少量多次洗涤锥形瓶或高脚烧杯,洗液合并于容量瓶中并定容至刻度,混匀备用;同时作试剂空白.6.3 测定6.3.1 仪器条件:根据各自仪器性能调至最佳状态.参考条件为波长283。

食品中镉的测定方法.doc 食品中镉的测定方法一、引言镉是一种广泛存在于环境中的重金属元素,它具有高毒性和慢性积累的特点。
人体摄入过多的镉会导致肾脏损害、骨质疏松、癌症等健康问题。
因此,对食品中镉的测定具有重要的意义。
二、常用的测定方法1. 原子吸收光谱法原子吸收光谱法是一种广泛应用于食品中金属元素测定的方法。
该方法通过测量样品中镉吸收光谱的强度来定量分析镉的含量。
该方法具有准确性高、灵敏度好的特点,但需要使用昂贵的仪器设备。
2. 电感耦合等离子体质谱法电感耦合等离子体质谱法是一种高灵敏度、高选择性的分析方法,对于微量元素的测定具有独特的优势。
该方法通过将样品中的镉离子转化为气态镉离子,并通过质谱仪测量镉离子的质量来定量分析镉的含量。
该方法具有高灵敏度、高准确性的特点,但需要昂贵的仪器设备和专业的操作技术。
3. 电感耦合等离子体发射光谱法电感耦合等离子体发射光谱法是一种常用的金属元素分析方法。
该方法通过将样品中的镉转化为气态镉原子,并利用光谱仪测量镉原子的发射光谱来定量分析镉的含量。
该方法具有高准确性、高灵敏度的特点,但需要使用昂贵的仪器设备。
4. 亚硝胺分析法亚硝胺分析法是一种常用的食品中有机镉测定方法。
该方法通过将样品中的有机镉转化为亚硝胺,再利用特定的化学反应将亚硝胺转化为可测定的产物,并通过光谱仪等设备测量产物的吸光度来定量分析镉的含量。
该方法简单、灵敏度高,但对样品的预处理要求较高。
三、结论食品中镉的测定方法有多种,其中原子吸收光谱法、电感耦合等离子体质谱法、电感耦合等离子体发射光谱法和亚硝胺分析法是较为常用的方法。
不同的方法有各自的特点和适用范围,选择合适的方法需要考虑样品的特性、测定的灵敏度和准确性要求以及实验条件等因素。
食品科技当前铝被公认为食品污染物,国家需在考虑食品中铝本底含量和风险评估基础上,制定各类食品中铝含量限值。
除了食品生产企业需要合理添加含铝添加剂外,国家也需加快完善食品安全标准,实现与国际标准的接轨。
科学应用现行标准检测方法,依据各类食品含铝本底值和各类检测方法的检出限科学选择检测方法,准确判别食品中铝元素含量。
1 食品中铝元素对健康的危害性1.1 食品中铝元素的来源首先是环境中天然存在的铝元素。
由于铝是地壳中含量最高的金属,因此在日常饮水、食物中会存在一定的铝元素。
其次是使用的烹饪用具和锡纸中含有铝元素。
据调查显示,铝制食品器具中,铝的溶出量相对较高,长期使用和洗刷会导致不粘涂层脱落,烹煮酸性食物也会加大食品中的铝含量。
相对而言,陶瓷器具和玻璃器具等的铝溶出量更低。
最后,来源于含铝食品添加剂,当前常见的有膨松剂、铵明矾和钾明矾等。
由于明矾的絮凝效果较好,民间常用其作为净水剂。
此外,一些淀粉类食物中也会加入明矾,从而让食物更加劲道,这样一来就会导致食品中存在大量的铝元素残留[1]。
1.2 铝对人体的危害性人体中铝元素含量超标,会严重损坏人体健康。
铝虽然不会导致人体急性中毒,但超过一定值也会产生危害,人体在摄入铝之后可以自行将其中的10%~15%排出体外,而大部分积蓄在体内,通过与各类蛋白质和酶的结合对体内多种生化反应产生影响。
因此,长期摄入铝元素,会损坏人的大脑,尤其对孕妇、老人和小孩的影响最大,容易引发儿童发育迟缓、老年痴呆等问题[2]。
2 食品中铝元素的测定方法与应用2.1 前处理方法2.1.1 湿法消解在GB 5009.182-2017《食品安全国家标准食品中铝的测定》中,对湿法消解进行了规定。
湿法处理虽然有机物分解速度快、元素损失少、处理时间短,但在试剂的使用量较大,消解过程中还会产生大量有害气体,同时使用过程需要人随时观察照看。
2.1.2 干法消解干法消解需保证样品的完全碳化,否则容易着火引发重大损失。