primer5和Oligo结合设计引物步骤及心得
- 格式:docx
- 大小:55.33 KB
- 文档页数:5

作为目前最好、最专业的引物设计软件,Oligo的功能很强,在这里我们介绍它的一些主要功能:如:普通引物对的搜索、测序引物的设计、杂交探针的设计以及评估引物对质量等等。
在正式进行引物设计前,我们首先面临的一个任务就是向Oligo程序导入模板序列,根据不同的实验情况,导入模板有三种方法:1, 直接用键盘输入:a, 点击file菜单中的New Sequence 浮动命令,或直接点击工具栏中的New Sequence 命令,进入序列展示窗口;b,此时即可键入DNA序列;c,如果需要的话,Oligo提供碱基回放功能,在边键入时边读出碱基,防止输入错误.点击Edit菜单中的“Readback on"即可.2,利用复制和粘贴:当我们序列已经作为TXT文件存在或其它oligo不能直接open的文件格式,如word文件。
html格式,这个功能就显得很有用了。
在相应文件中复制序列后在序列展示窗口粘贴,oligo会自动去除非碱基字符。
当序列输入或粘贴完成后,点击Accept/Discard菜单中的Accept浮动命令,即可进入引物设计模式。
3,如果序列已经保存为Seq格式或者FASTA,GenBank格式时,oligo就可以直接打开序列文件.点击File菜单中的“Open”浮动命令,找到所需文件,打开即可.进入引物设计模式后,oligo一般会弹出三个窗口,分别是6-碱基频率窗口,碱基退火温度窗口以及序列内部碱基稳定性窗口,其中的退火温度窗口是我们引物设计的主窗口,其它的两个窗口则在设计过程中起辅助作用,比如6-碱基频率窗口可以使我们很直观地看到所设计引物在相应物种基因组中的出现频率,如果我们的模板是基因组DNA或混合DNA时,该信息就显得有用了,而内部稳定性窗口则可以显示引物的5’端稳定性是否稍高于3’端等.一,普通引物对的搜索:以Mouse 4E(cDNA序列)为例。
我们的目的是以Mouse 4E(2361 bp)为模板,设计一对引物来扩增出600-800bp长的pcr产物。

PCR引物设计知识与技巧分析PCR引物设计知识与技巧分析Posted on 30 五月2009 by 柳城,阅读526 简洁版繁體自从1985年Mullis等发明了具有划时代意义的聚合酶链式反应(PCP) 以来,PCR已经成为了分子生物学领域最基本也是最重要的技术手段之一。
然而能否找到一对合适的核苷酸片段作为引物,使其有效地扩增模板DNA序列,无疑决定着PCR的成败。
现在动物遗传育种早已进入了分子时代,在基因水平寻求影响动物遗传表型的新基因突显重要,因此引物设计无疑又成为了寻找新基因的重中之重。
1 引物的设计以及初步筛选引物的设计与初步筛选基本上通过一些分子生物学软件和相关网站来完成的,目前运用软件Primer Premier 5 或美国whitehead 生物医学研究所基因组研究中心在因特网上提供的一款免费在线PCR引物设计程序Primer 3来设计引物,再用软件Oligo 6进行引物评估,就可以初步获得一组比较满意的引物。
但是对于初学者来说,运用软件和程序来设计引物好象无从着手,其实只要我们掌握了引物设计的基本原则和注意事项,所有问题便迎刃而解。
因为无论是软件还是程序,都是以这些基本原则和注意事项为默认标准来进行引物设计的。
所以,我们在进行引物设计的时候大可不必在软件和程序的参数上花费过多的时间来思考,如果没有特殊要求我们完全可以把一些参数设为默认值。
下面我们主要讨论一下引物设计的原则和注意事项。
①引物的长度一般为15-30 bp,最好在18~24 bp,因为太短易形成错配(False priming) 降低特异性,而太长也会降低特异性,并且降低产量。
②引物在模板内最好具有单一性,也就是说在模板内部没有错配。
特别是3’端,一定要避免连续4个以上的碱基互补错配。
③引物序列的GC 含量最好在40%一60%,且上下游引物序列GC含量的差异不要太大,3’端最后5个碱基最好不要富含GC,特别是连续3个的G或C。

一,普通引物对的搜索:以Mouse 4E(cDNA序列)为例。
我们的目的是以Mouse 4E(2361 bp)为模板,设计一对引物来扩增出600-800bp长的PCR产物。
1,点击“Search“菜单中的”For Primers and Probes“命令,进入引物搜索对话框;2,由于我们要设计的是一对PCR引物,因此正、负链的复选框都要选上,同时选上Compatible pairs。
在Oligo默认的状态下,对此引物对的要求有:a,无二聚体;b,3’端高度特异,GC含量有限定,d,去除错误引发引物等。
3,剩下的工作是确定上、下游引物的位置及PCR产物的长度以及引物设计参数。
①单击:“search Ranges”按钮,弹出“Search Ranges”对话框。
输入上游引物的范围:1-2000,下游引物的位置:100-2300;PCR产物的长度600-800bp。
②单击“Paramaters”按钮进入“Search Parameters”对话框,对话框种分三个活页,分别是:不同设定,参数以及更多参数。
③在“普通设定”窗口,为我们提供了对引物非常直观的设定方法,从高到低分六个等级,最后还有一个用户定制选项。
④当我们对引物的各种参数的含义及应该设定多大值并不是特别清楚时,就可以直接设定Very high/High等来完成对引物设计参数的设定。
⑤当我们选中“Automatically Change String”后,Oligo会在引物搜索过程中:如果在高等级设定中无法找到引物对时自动降低一个定级来进行搜索,知道找到引物对。
在设计反向PCR引物对时,就选中“Inverse PCR”复选框。
⑥我们还可以让引物的长度可以改变,以适应设定的Tm值或PE?(Prime Effitions,引发效率)。
也可以限定所选引物对的最大数目。
⑦在“Parameters”窗口中,实际上需要我们改动的只有引物的长度,根据试验的要求作相应的改变,如23nt。

一、引物设计step by step1、在NCBI上搜索到目的基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
2、用Primer Premier5搜索引物①打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
②此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。
在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200b p的产物电泳跑得较散,所以可以选择300~500bp.③点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
④对于引物的序列,可以简单查看一下,避免出现下列情况:3’不要出现连续的3个碱基相连的情况,比如GGG或CCC,否则容易引起错配。
此窗口中需要着重查看的包括:T m应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的T m值最好不要相差太多,大概在2度以下较好。
该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。
若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好。
但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。
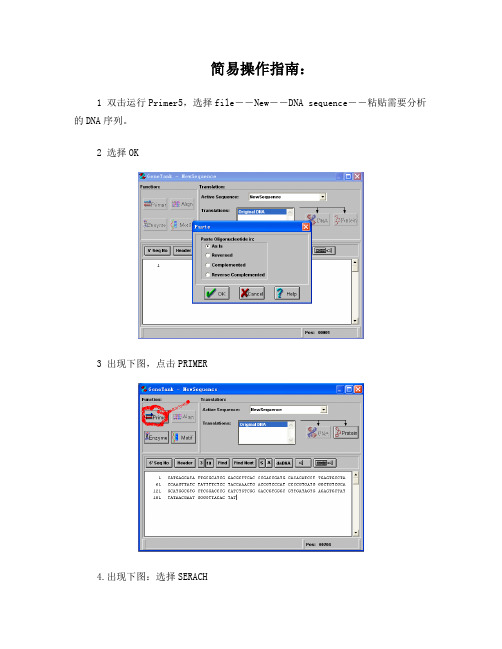
简易操作指南:
1 双击运行Primer5,选择file--New――DNA sequence――粘贴需要分析的DNA序列。
2 选择OK
3 出现下图,点击PRIMER
4.出现下图:选择SERACH
5.出现下图,定义正向引物大概位置和反义引物大概位置,引物片段的大小以及目标片段的大小的相关参数以及成对设计还是单独正向或反向设计。
如下设置
点击OK后
再点击OK。
点击每一条目就会出现,该引物对的相关信息与评价
点击S为正向引物,A为反向引物;
Hairpin:为发卡结构,指一条引物自身配对
Dimer:二聚体,为同向两引物之间配对
False dimmer:指引物与DNA序列的其他地方有配对情况Cross Dimer:正向与反向引物之间发生错配。
如感觉满意,则可以点击Edit Primer 将正向和反向引物选用。

简述引物设计流程及结果英文回答:Primer design is a crucial step in many molecular biology experiments, including PCR, qPCR, and DNA sequencing. It involves designing short DNA sequences, known as primers, that specifically bind to the target DNA region and initiate DNA synthesis. The primer design process typically consists of several steps, including target sequence identification, primer design software selection, primer design parameters setting, primer specificity checking, and primer optimization.The first step in primer design is to identify the target DNA sequence that needs to be amplified or analyzed. This could be a specific gene or a region of interest in the genome. Once the target sequence is identified, the next step is to select a primer design software. There are several software tools available, such as Primer3, OligoAnalyzer, and NCBI Primer-BLAST, which can assist indesigning primers based on various parameters.After selecting the primer design software, the next step is to set the primer design parameters. These parameters include primer length, melting temperature (Tm), GC content, and potential secondary structures. The primer length is usually between 18-25 nucleotides, with 20 nucleotides being the most commonly used length. The Tm is the temperature at which half of the primers are bound to the target DNA sequence, and it is typically set to be around 55-65°C. The GC content is the percentage of guanine (G) and cytosine (C) bases in the primer sequence, and it is usually set to be around 40-60%. Potential secondary structures, such as hairpins or primer dimers, should also be avoided.Once the primer design parameters are set, the next step is to check the specificity of the primers. This can be done using the primer design software or by performing a BLAST search against the target DNA sequence. The primers should only bind to the intended target sequence and not to any other regions in the genome. This is important toensure accurate and specific amplification or analysis ofthe target DNA.After checking the specificity, the final step isprimer optimization. This involves adjusting the primer design parameters, such as the Tm and GC content, toimprove the efficiency and specificity of the primers. It may also involve designing multiple primer pairs andtesting them experimentally to determine the bestperforming primers.The result of the primer design process is a set of primers that are specific to the target DNA sequence and have optimal design parameters. These primers can then be used in various molecular biology experiments, such as PCR, qPCR, and DNA sequencing, to amplify or analyze the target DNA.中文回答:引物设计是许多分子生物学实验中的一个关键步骤,包括PCR、qPCR和DNA测序。
引物设计的标准操作规程(编号:004)1、软件使用1.1 推荐软件:Primer Premier 5.01.2 优点:操作简单、显示各种参数改变和二聚体、异二聚体、发夹结构等。
1.3 本地同类软件:DNAClub;Oligo 6.22;Vector NTI Suit;Dnasis;Omiga;Dnastar;DNAMAN (Lynnon Biosoft, Quebec, Canada)。
1.4 网上同类软件:Primer3、JaMBW(European Molecular Biology Laboratory of Heidelberg 开发)。
http://210.72.11.60网站已引进并调试好这两种软件。
2、推荐操作引物搜索:Primer Premier 5.0、引物评价:Oligo 6.223、引物设计的原则首先引物要跟模板紧密结合,其次引物与引物之间不能有稳定的二聚体或发夹结构存在,再次引物不能在别的非目的位点引起DNA聚合反应(即错配)。
围绕这几条基本原则,设计引物需要考虑诸多因素,如引物长度(primer length)、产物长度(product length)、序列Tm值(melting temperature)、ΔG值(internal stability)、引物二聚体及发夹结构(duplex formation and hairpin)、错误引发位点(false priming site)、引物及产物GC 含量(composition),有时还要对引物进行修饰,如增加限制酶切点,引进突变等。
以使用Oligo 软件分析设计引物为例,笔者总结出以下的要点:3.1 引物的长度一般为18-25bp,引物长度过长会导致延伸温度过高,从而影响DNA聚合酶的效率,上下游引物长度差别最好不要大于3bp。
3.2 引物最好在模板DNA的保守区内设计。
DNA序列的保守区是通过物种间相似序列的比较确定的。
第一贴记得当时搞引物设计的时候,还是我的大师兄领入门的,在此谢过了。
师兄教了一些基本原则后,就放羊了。
接着自己捣鼓来捣鼓去,在导师的实验室内设计了十几条引物,用起来还蛮好的,逐渐赚了点名气。
后来,就和上海鼎安合作了,帮别人设计引物在他们公司订购引物,拿点回扣。
别人拿给我设计的引物,都是很头痛。
因为客户往往都是自己捣鼓一下,不行了才到我这。
到现在已经成功设计了四五十对引物了。
不过至今money颗粒无收,就不了了之,主要是客户拖欠的问题,不过不后悔,毕竟是不错的经历啊。
所以,也算是有点个人观点,和大家分享,不对的地方请指正。
我用Primer 5.0搜索引物、Oligo 6.0评估引物,这是根据各自所长定的。
但有个朋友,他就只用Oligo,说设计的引物也还可以。
Primer 5.0搜索引物:1.Primer Length我常设置在18-30bp,短了特异性不好,长了没有必要。
当然有特殊要求的除外,如加个酶切位点什么的。
2.PCR Product size最好是100-500bp之间,小于100bp的PCR产物琼脂糖凝胶电泳出来,条带很模糊,不好看。
至于上限倒也不必要求苛刻。
3.Search parameters还是选Manual吧,Search stringency应选High,GC含量一般是40-60%。
其它参数默认就可以了。
4.搜索出来的引物,按Rating排序,逐个送Oligo软件里评估。
当然,搜索出的引物,其扩增产物很短,你可以不选择它,或是引物3端≥2个A或T,或引物内部连续的G或C太多,或引物3端≥2个G或C,这样的引物应作为次选,没得选了就选它。
对于这样的引物,如果其它各项指标还可以,我喜欢在引物末端去掉一个不满意的或加上一个碱基,看看引物的评估参数有没有变好点。
Oligo 6.0评估引物:1.在analyze里,Duplex Formation不管是上游引物、下游引物还是上下游引物之间,The most stable 3’-Dimer绝对值应小于4.5kcal/mol, The most stable Dimer overall绝对值一般应小于多少kcal/mol跟PCR退火温度有关,我几次实验感觉在PCR退火温度在65°的时候,The most stable Dimer overall 6.7kcal/mol没有问题。
引物设计step by step1、在NCBI上搜索到目的基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
2、用Primer Premier5搜索引物①打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
②此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。
在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择300~500bp.③点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
④对于引物的序列,可以简单查看一下,避免出现下列情况:3’不要出现连续的3个碱基相连的情况,比如GGG或CCC,否则容易引起错配。
此窗口中需要着重查看的包括:Tm 应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。
该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。
若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好。
但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。
Primer5.0 目的基因序列引物设计
GA TTtCTTGGCTTtATA TA TCTTGT GGAaAGGaCGAAACACCGTGCTCGCTTCGGCAGCAC ATATACTAGTCGACGGGTCTAGACAA TGA TGCTGGGTAA TGACACCAAGCTGGGACTG GTACAGAAAGTCAGAGAACACTTACAGAACGGCA TCTAGACAATGA TGCTGGGTAATA CACTTACAGAACGGCA TCTAGA TGCCGTTCTGTAAGTGTTTGTTGAATGAATGAGTGTT GAACAAACTGCTAAGGTATCTTT ACAAGGTAG
从中扩增出目的基因片段(绿色的部分):
首先:将绿色部分复制到primer5.0引物设计软件中,ctrl+v用鼠标右键不管用
.选择As is后,惦记OK出现:
然后惦记,出现
图中右上角标记,其中惦记S合成的是上游引物,A是下游引物,点击S后出现,然后点击出现
这些就是上游引物,选中后ctrl+c复制到引物合成单中,发给公司,下游引物:回到
点击A出现
用前后调节框,如下,将序列向右移动移到最后,得到下图
注意显示的是3—5,ctrl+c—ctrl+v后悔发现序列变成5---3了,这是软件的好处,因为公司引物合成就是从5—3的
其实上下游引物只是相对的,将得到的引物序列填表后发给公司就好了。
primer5和Oligo结合设计引物步骤及心得 来源:北京力途科技有限公司 2010-5-21 访问量:5404 评论(0) 分享1
一、引物设计step by step
1、在NCBI上搜索到目的基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
2、用Primer Premier5搜索引物 ①打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。点击Primer,进入引物窗口。
②此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。在Search Parameters里面,可以设定相应参数。一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择 300~500bp.
③点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
④对于引物的序列,可以简单查看一下,避免出现下列情况: 3’不要出现连续的3个碱基相连的情况,比如GGG或 CCC,否则容易引起错配。此窗口中需要着重查看的包括:Tm应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好。但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。对于引物具体详细的评价需要借助于Oligo来完成,Oligo自身虽然带有引物搜索功能,但其搜索出的引物质量感觉不如Primer5. ⑤在Primer5窗口中,若觉得某一对引物合适,可以在搜索结果窗口中,点击该引物,然后在菜单栏,选择File-Print-Current pair,使用PDF虚拟打印机,即可转换为Pdf文档,里面有该引物的详细信息。
3、用Oligo验证评估引物 ①在Oligo软件界面,File菜单下,选择Open,定位到目的cDNA序列(在primer中,该序列已经被保存为Seq文件),会跳出来两个窗口,分别为Internal Stability(Delta G)窗口和Tm窗口。在Tm窗口中,点击最左下角的按钮,会出来引物定位对话框,输入候选的上游引物序列位置(Primer5已经给出)即可,而引物长度可以通过点击Change-Current oligo length来改变。定位后,点击Tm窗口的Upper按钮,确定上游引物,同样方法定位下游引物位置,点击Lower按钮,确定下游引物。引物确定后,即可以充分利用Analyze菜单中各种强大的引物分析功能了。
②Analyze中,第一项为Key info,点击Selected primers,会给出两条引物的概括性信息,其中包括引物的Tm值,此值Oligo是采用nearest neighbor method计算,会比Primer5中引物的Tm值略高,此窗口中还给出引物的Delta G和3’端的Delta G.3’端的Delta G过高,会在错配位点形成双链结构并引起DNA聚合反应,因此此项绝对值应该小一些,最好不要超过9。
③Analyze中第二项为Duplex Formation,即二聚体形成分析,可以选择上游引物或下游引物,分析上游引物间二聚体形成情况和下游引物间的二聚体情况,还可以选择Upper/Lower ,即上下游引物之间的二聚体形成情况。引物二聚体是影响PCR反应异常的重要因素,因此应该避免设计的引物存在二聚体,至少也要使设计的引物形成的二聚体是不稳定的,即其Delta G值应该偏低,一般不要使其超过4.5kcal/mol,结合碱基对不要超过3个。Oligo此项的分析窗口中分别给出了3’端和整个引物的二聚体图示和Delta G值。
④Analyze中第三项为Hairpin Formation,即发夹结构分析。可以选择上游或者下游引物,同样,Delta G值不要超过4.5kcal/mol,碱基对不要超过3个。
Analyze中第四项为Composition and Tm,会给出上游引物、下游引物和产物的各个碱基的组成比例和Tm值。上下游引物的GC%需要控制在40%~60%,而且上下游引物之间的GC%不要相差太大。Tm值共有3个,分别采用三种方法计算出来,包括nearest neighbor method、%GC method和2(A+T)+4(G+C)method,最后一种应该是Primer5所采用的方法,Tm值可以控制在50~70度之间。
第五项为False Priming Sites,即错误引发位点,在Primer5中虽然也有False priming分析,但不如oligo详细,并且oligo会给我正确引发效率和错误引发效率,一般的原则要使误引发效率在100以下,当然有时候正确位点的引发效率很高的话,比如达到400~500,错误引发效率超过100幅度若不大的话,也可以接受。
⑤Analyze中,有参考价值的最后一项是“PCR”,在此窗口中,是基于此对引物的PCR反应Summary,并且给出了此反应的最佳退火温度,另外,提供了对于此对引物的简短评价。若该引物有不利于PCR反应的二级结构存在,并且Delta G值偏大的话,Oligo在最后的评价中会注明,若没有注明此项,表明二级结构能值较小,基本可以接受。
⑥引物评价完毕后,可以选择File-Print,打印为PDF文件保存,文件中将会包括所有Oligo软件中已经打开的窗口所包括的信息,多达数页。因此,打印前最好关掉Tm窗口和Delta G窗口,可以保留引物信息窗口、二级结构分析窗口(若存在可疑的异常的话)和PCR窗口。
4、引物确定后,对于上游和下游引物分别进行Blast分析,一般来说,多少都会找到一些其他基因的同源序列,此时,可以对上游引物和下游引物的blast结果进行对比分析,只要没有交叉的其他基因的同源序列就可以。
二、引物设计过程中的心得 1、Primer 5.0搜索引物 ①Primer Length我常设置在18-30bp,短了特异性不好,长了没有必要。当然有特殊要求的除外,如加个酶切位点什么的。
②PCR Product size最好是100-500bp之间,小于100bp的PCR产物琼脂糖凝胶电泳出来,条带很模糊,不好看。至于上限倒也不必要求苛刻。
③Search parameters还是选Manual吧,Search stringency应选High,GC含量一般是40-60%。其它参数默认就可以了。
④搜索出来的引物,按Rating排序,逐个送Oligo软件里评估。当然,搜索出的引物,其扩增产物很短,你可以不选择它,或是引物3端≥2个A或T,或引物内部连续的G或C太多,或引物3端≥2个G或C,这样的引物应作为次选,没得选了就选它。对于这样的引物,如果其它各项指标还可以,我喜欢在引物末端去掉一个不满意的或加上一个碱基,看看引物的评估参数有没有变好点。
2、Oligo 6.0评估引物 ①在analyze里,Duplex Formation不管是上游引物、下游引物还是上下游引物之间,The most stable 3’-Dimer绝对值应小于4.5kcal/mol, The most stable Dimer overall绝对值一般应小于多少kcal/mol跟PCR退火温度有关,我几次实验感觉在PCR退火温度在65°的时候,The most stable Dimer overall 6.7kcal/mol没有问题。
②Hairpin Formation根据黄金法则 ③False priming sites: Primer的priming efficiency应该是错配地方的4倍左右,更多当然更好。
④在PCR栏,设计者感觉其所显示的optimal annealing temperature数值值得参考。在PCR摸索条件的时候,退火温度为其数值加减2的范围就可以了。
⑤Internal stability很重要:我们希望引物的内部稳定性是中间高、两边低的弧形,最起码保证3端不要过于稳定。下图引物3端过于稳定,很容易导致不适当扩增。△G参照黄金法则,这其实很好理解:把一滴水放到大海里,这滴水就会不停的扩散分布,扩散的越厉害越稳定,所以△G绝对值越大结构越稳定。
3、其他 ①两个评价系统不一样,丁香园战友感觉oligo评价引物好点,primer出来的引物,一般按效率排序,再结合退火温度和引物长度,选择引物到oligo测试。这是初步的选择,其实引物到了oligo里,退火温度也不一样。
②3端的二聚体应该避免,这个要看退火温度决定,一个50°的退火温度肯定和65°对二聚体的影响不一样了,一般来讲尽量控制在-4.5kcal/mol以下(丁香园战友观点,很多东西真得还是需要自己摸索)。
③我们感觉3端有A无A影响不大,3端有T是不是一定不行,不见得。软件是评估,法则也不是没有例外,不是1+1=2那么确定。
④错配和二聚体谁轻谁重,丁香园战友觉得“到致命的程度”谁都重要,在设计的时候,尽量两个都不得罪。
⑤GC含量并非不重要,它直接影响引物各端稳定性,3端来两个G或C,稳定性就上去了,粘在模板上很牢。所以设计引物的时候,会尽量避免这样的情况出现。