引物设计基本方法
- 格式:doc
- 大小:39.00 KB
- 文档页数:4

2、引物长度一般在15-30碱基之间。
引物长度(primer length)常用的是18-27bp,但不应大于38bp,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。
3、引物GC含量在40%~60%之间,Tm值最好接近72℃。
GC含量(composition)过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。
有效启动温度,一般高于Tm 值5-10℃。
若按公式Tm=4(G+C+2(A+T)估计引物的Tm值,则有效引物的Tm为55-80℃,其Tm值最好接近72℃以使复性条件最佳。
4、引物3'端要避开密码子的第3位。
如扩增编码区域,引物3'端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。
5、引物3'端不能选择A,最好选择T。
引物3'端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3'端最好选择T。
6、碱基要随机分布。
引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。
降低引物与模板相似性的一种方法是,引物中四种碱基的引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。
这种二级结构会因空间位阻而影响引物与模板的复性结合。
引物自身不能有连续4个碱基的互补。
两引物之间也不应具有互补性,尤其应避免3'端的互补重叠以防止引物二聚体(Dimer 与Cross dimer)的形成。
引物之间不能有连续4个碱基的互补。
引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal /mol)。

设计引物的方法
设计引物的方法主要包括以下步骤:
1. 获取基因序列:可以通过NCBI等网站获取基因序列,或者通过实验测序得到基因序列。
2. 设置引物参数:包括引物长度、Tm值、GC含量等。
引物长度一般在
18-30bp之间,Tm值在55-65℃之间,GC含量在30%-60%之间。
此外,还需要考虑引物的特异性,避免与基因组中的其他序列发生非特异性结合。
3. 选择合适的引物设计软件:例如Primer Premier、Oligo、Primer3等引物设计软件,这些软件可以根据设定的参数和基因序列自动设计引物。
4. 验证引物:通过BLAST等工具对设计的引物进行验证,确保引物特异性。
5. 合成引物:将验证合格的引物合成出来,用于后续的实验。
需要注意的是,引物设计需要一定的专业知识和经验,建议在专业人士的指导下进行。

设计引物的方法
首先,确定目标序列是进行引物设计的第一步。
目标序列的选
择应该考虑到所需扩增的DNA片段的长度和特点,以及引物的位置
和方向。
在确定了目标序列之后,就可以根据目标序列的特点来选
择合适的引物设计方法了。
一种常用的引物设计方法是基于序列比对的方法。
通过对目标
序列与相关物种的序列进行比对,可以找到共同的保守区域,从而
设计出具有较高特异性的引物。
这种方法适用于目标序列与已知序
列高度保守的情况,可以有效地提高引物的特异性和扩增效率。
另一种常用的引物设计方法是基于引物特性的方法。
在这种方
法中,可以根据引物的特性来设计引物,例如引物的长度、GC含量、熔解温度等。
通过调整这些引物特性,可以提高引物的特异性和扩
增效率。
同时,还可以利用一些引物设计软件来辅助设计引物,这
些软件可以根据一定的算法和模型来预测引物的性能,提高引物设
计的准确性和效率。
除了以上介绍的两种方法外,还有一些其他的引物设计方法,
例如基于结构的方法、基于引物库的方法等。
这些方法都有各自的
优缺点,可以根据实际情况来选择合适的方法进行引物设计。
总的来说,设计引物的方法是一个复杂而又重要的过程。
在进行引物设计时,需要考虑到目标序列的特点、引物的特性以及实验条件等多个因素。
通过选择合适的引物设计方法,可以设计出具有较高特异性和扩增效率的引物,从而提高PCR扩增和测序结果的准确性和可靠性。
希望本文介绍的引物设计方法能够对研究人员在实验中有所帮助。
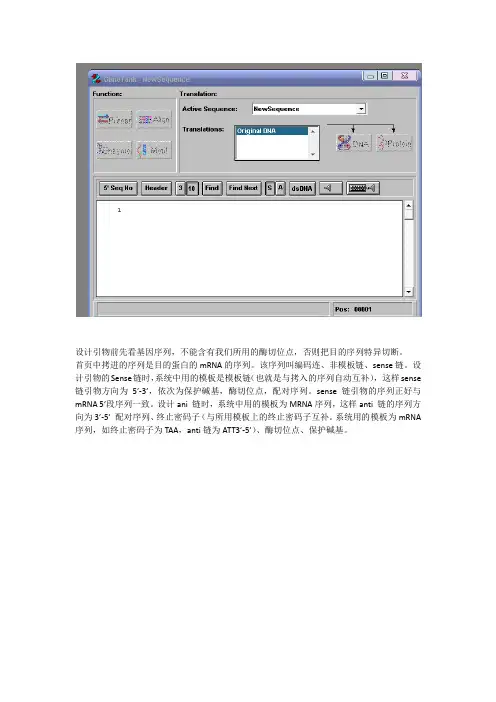
设计引物前先看基因序列,不能含有我们所用的酶切位点,否则把目的序列特异切断。
首页中拷进的序列是目的蛋白的mRNA的序列。
该序列叫编码连、非模板链、sense链。
设计引物的Sense链时,系统中用的模板是模板链(也就是与拷入的序列自动互补),这样sense 链引物方向为5’-3’,依次为保护碱基,酶切位点,配对序列。
sense链引物的序列正好与mRNA 5’段序列一致。
设计ani 链时,系统中用的模板为MRNA序列,这样anti 链的序列方向为3’-5’配对序列、终止密码子(与所用模板上的终止密码子互补。
系统用的模板为mRNA 序列,如终止密码子为TAA,anti链为ATT3’-5’)、酶切位点、保护碱基。


引物设计(特别有用,良心推荐)引物篇1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。
DNA合成仪有很多种, 主要都是由ABI/PE 公司生产,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。
亚磷酰胺三酯法合成DNA片段,具有高效、快速的偶联以及起始反应物比较稳定的特点。
亚磷酰胺三酯法是将DNA固定在固相载体上完成DNA链的合成的,合成的方向是由待合成引物的3'端向5'端合成的,相邻的核苷酸通过3'→5'磷酸二酯键连接。
第一步是将预先连接在固相载体CPG上的活性基团被保护的核苷酸与三氯乙酸反应,脱去其5'-羟基的保护基团DMT,获得游离的5'-羟基;第二步,合成DNA的原料,亚磷酰胺保护核苷酸单体,与活化剂四氮唑混合,得到核苷亚磷酸活化中间体,它的3'端被活化,5'-羟基仍然被DMT保护,与溶液中游离的5'-羟基发生缩合反应。
第三步,带帽(capping)反应,缩合反应中可能有极少数5'-羟基没有参加反应(少于2%),用乙酸酐和1-甲基咪唑终止其后继续发生反应,这种短片段可以在纯化时分离掉。
第四步,在氧化剂碘的作用下,亚磷酰形式转变为更稳定的磷酸三酯。
经过以上四个步骤,一个脱氧核苷酸被连接到固相载体的核苷酸上。
再以三氯乙酸脱去它的5'-羟基上的保护基团DMT,重复以上步骤,直到所有要求合成的碱基被接上去。
合成过程中可以观察TCA处理阶段的颜色判定合成效率。
通过氨水高温处理,连接在CPG上的引物被切下来,通过OPC, PAGE等手段纯化引物,成品引物用C18浓缩,脱盐,沉淀。
沉淀后的引物用水悬浮,测定OD260定量,根据定单要求分装。
2.引物纯化方式有哪些,如何选择?◆C18柱脱盐:有人称其为简易反相柱,它对DNA有特异性的吸附,可以被有机溶解洗脱,但不会被水洗脱,所以能有效地去除盐分。

Primer 5.0搜索引物:1.Primer Length我常设置在18-30bp,短了特异性不好,长了没有必要。
当然有特殊要求的除外,如加个酶切位点什么的。
2.PCR Product size最好是100-500bp之间,小于100bp的PCR产物琼脂糖凝胶电泳出来,条带很模糊,不好看。
至于上限倒也不必要求苛刻。
3.Search parameters还是选Manual吧,Search stringency应选High,GC含量一般是40-60%。
其它参数默认就可以了。
4.搜索出来的引物,按Rating排序,逐个送Oligo软件里评估。
当然,搜索出的引物,其扩增产物很短,你可以不选择它,或是引物3端≥2个A或T,或引物内部连续的G或C太多,或引物3端≥2个G或C,这样的引物应作为次选,没得选了就选它。
对于这样的引物,如果其它各项指标还可以,我喜欢在引物末端去掉一个不满意的或加上一个碱基,看看引物的评估参数有没有变好点。
Oligo 6.0评估引物:1.在analyze里,Duplex Formation不管是上游引物、下游引物还是上下游引物之间,The most stable 3’-Dimer绝对值应小于4.5kcal/mol, The most stable Dimer overall绝对值一般应小于多少kcal/mol跟PCR退火温度有关,我几次实验感觉在PCR退火温度在65°的时候,The most stable Dimer overall 6.7kcal/mol没有问题。
2.Hairpin Formation根据黄金法则3.False priming sites: Primer的priming efficiency应该是错配地方的4倍左右,更多当然更好。
4.在PCR栏,个人感觉其所显示的optimal annealing temperature数值值得参考。
在PCR摸索条件的时候,退火温度为其数值加减2的范围就可以了。

设计引物的方法
首先,我们需要明确引物的特性。
引物是一种短链的DNA或RNA序列,通常用于PCR扩增、实时荧光定量PCR、测序等实验中。
因此,引物的选择需要考虑
到引物的长度、GC含量、配对温度等特性。
一般来说,引物的长度应在18-25个
碱基对之间,GC含量应在40%-60%之间,配对温度应在50-65摄氏度之间。
这些
特性的选择将直接影响到引物的扩增效率和特异性。
其次,针对目标序列的特点,我们需要选择合适的引物设计方法。
对于已知的
序列,我们可以利用计算机软件进行引物设计,如Primer3、Beacon Designer等。
这些软件可以根据用户输入的序列信息,自动给出符合要求的引物设计方案。
对于未知序列,我们可以采用引物库筛选的方法,从引物库中挑选符合要求的引物进行实验。
此外,实验的具体要求也是引物设计的考量因素之一。
在进行引物设计时,我
们需要考虑到实验的目的、样本的特点、实验条件等因素。
比如,在进行实时荧光定量PCR实验时,我们需要选择特定的引物和探针,以确保实验的准确性和灵敏度。
在进行测序实验时,我们需要选择特定的引物,以确保测序结果的可靠性和准确性。
综上所述,设计引物的方法需要考虑引物的特性、目标序列的特点以及实验的
具体要求。
在进行引物设计时,我们可以利用计算机软件进行设计,也可以采用引物库筛选的方法。
同时,我们需要根据实验的具体要求,选择合适的引物设计方案。
希望以上内容能够帮助大家更好地进行引物设计,提高实验的效率和准确性。

简述pcr引物设计的基本步骤
PCR引物设计是PCR技术中至关重要的一步,它直接影响到PCR 反应的特异性和效率。
以下是PCR引物设计的基本步骤:
1. 确定目标序列,首先需要确定要扩增的目标DNA序列,这可以是基因、片段或者其他特定的DNA区域。
2. 引物长度,一般来说,PCR引物的长度应在18-25个碱基对之间,太短会影响特异性,太长则会影响引物的合成效率。
3. 引物的GC含量,引物的GC含量应在40-60%之间,这有助于提高引物与模板DNA的亲和力。
4. 引物特异性,引物应该与目标DNA序列高度特异性地结合,避免与其他非特异性DNA结合。
5. 引物序列的避让,避免引物序列中出现相互补的碱基对,以免引物之间发生非特异性结合。
6. 引物的末端,引物的末端应该避免出现多余的碱基对,以免
影响PCR扩增的效率。
7. 引物的Tm值,引物的熔解温度(Tm值)应该相似,一般来说,它们之间的差异不应超过5摄氏度。
在进行PCR引物设计时,以上这些基本步骤可以帮助确保PCR 反应的特异性和效率。
同时,也可以利用一些生物信息学工具来辅助引物设计,如NCBI的Primer-BLAST、IDT的PrimerQuest等。
PCR引物设计的好坏直接关系到PCR扩增的成功与否,因此在实验前务必进行充分的设计和验证。

引物设计涉及的知识点引物设计是分子生物学和遗传学研究中的重要环节,它涉及到多个知识点和技术原理。
本文将系统地介绍引物设计所涉及的主要知识点,以帮助读者更好地理解和应用这一技术。
一、引物设计的基本原理在介绍引物设计涉及的具体知识点之前,我们先来了解一下引物设计的基本原理。
引物是DNA或RNA序列的一段寡聚核苷酸链,它具有与目标序列特异性互补的碱基配对能力。
引物设计的目的是在目标DNA或RNA序列上选择适当的引物,以确定需要扩增或检测的目标序列。
二、引物设计所涉及的知识点1. 碱基配对规则在引物设计中,了解DNA和RNA的碱基配对规则是必不可少的知识点。
DNA的碱基有腺嘌呤(A)、胸腺嘧啶(T)、鸟嘌呤(G)和胞嘧啶(C)四种,而RNA的碱基则将胸腺嘧啶(T)替换为尿嘧啶(U)。
根据碱基配对规则,A与T(或U)之间形成两个氢键,G与C之间形成三个氢键。
在引物设计中,准确把握碱基配对规则有助于合成特异性强的引物。
2. 目标序列的选择引物设计的首要任务是选择需要扩增或检测的目标序列。
在选择目标序列时,考虑到扩增效率和特异性,应优先选择长度适中(100-1000 bp)且具有一定GC含量的目标序列。
此外,还应避免选择含有较长重复序列或自身互补区域的目标序列,以避免引物间的非特异性结合。
3. 引物长度和GC含量的确定引物的长度和GC含量会直接影响引物的特异性和扩增效率。
一般来说,较短的引物(18-25 bp)具有较高的特异性和扩增效率,但在某些特殊情况下,如设计用于检测稀有突变的引物时,可以适当增加引物长度。
此外,引物的GC含量应在40%-60%之间,以保证引物和目标序列之间的热力学性质相近。
4. 引物的特异性评估在引物设计过程中,需要对设计的引物进行特异性评估,以确保其只与目标序列特异性结合。
常用的特异性评估方法包括BLAST分析和引物二聚体分析。
BLAST分析可以用于检查引物是否与非特定目标序列部分互补,而引物二聚体分析可以评估引物与自身及其他引物之间的非特异性结合情况。

microrna引物设计方法
miRNA引物设计是一种用于特定miRNA的PCR扩增的方法。
以下是一些miRNA引物设计的方法:
1. 基于序列同源性:该方法依据miRNA的序列同源性进行设计引物,即在miRNA序列中选择一小段高度保守的区域,并为该区域设计引物。
2. 基于互补性和mfold分析:该方法基于miRNA的互补性和稳定性进行设计引物。
通常,对于每个miRNA,在其5'和3'端分别设计2对引物,并通过mfold 软件预测引物结构和互补性,选择最适合的引物。
3. 基于基因组的位置:该方法首先在miRNA序列中找到相应的基因组位置,并分析它周围的DNA序列,以确定最佳的引物位置。
这种方法的优点是可以避免短读或多倍体问题。
4. 基于组合算法:该方法利用组合算法,计算miRNA序列中所有可能的引物序列。
然后,通过设计引物的各项指标,如长度、稳定性和互补性等,筛选最佳的引物序列。
通过以上的方法,可以设计出高效、可靠的miRNA引物,以用于qPCR和其他分子生物学分析。
PCR引物设计的黄金法则1. 引物最好在模板cDNA的保守区内设计。
DNA序列的保守区是通过物种间相似序列的比较确定的。
在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区。
2.引物长度一般在15~30碱基之间。
引物长度(primer length)常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。
3.引物GC含量在40%~60%之间,Tm值最好接近72℃。
GC含量(composition)过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。
有效启动温度,一般高于Tm值5~10℃。
若按公式Tm= 4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。
4.引物3′端要避开密码子的第3位。
如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。
5.引物3′端不能选择A,最好选择T。
引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3′端最好选择T。
6. 碱基要随机分布。
引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。
降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。
尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。
7. 引物自身及引物之间不应存在互补序列。
PCR引物设计汇总PCR(聚合酶链反应)引物是PCR反应中的两个核酸序列,它们分别位于待扩增的DNA片段的两端。
合理设计的PCR引物是PCR反应成功的关键,它们决定了PCR扩增的特异性和效率。
1.引物长度:一般选择18-25个碱基的引物长度。
引物过短可能导致非特异性扩增,引物过长则降低扩增效率。
2.引物碱基组成:引物中尽量避免使用连续的同类碱基,如连续的A、T、C或G。
同时,引物设计中应尽量均衡使用四种碱基,避免GC含量过高或过低。
3.引物Tm值:引物的Tm值(解链温度)是很重要的参数,它决定了PCR反应的温度条件。
一般,引物的Tm应在50-60摄氏度之间,且相互之间的Tm值差别不应超过两度。
4.引物特异性:引物应具有足够的特异性,以确保只扩增目标DNA片段,避免扩增到非特异性产物。
5.引物末端:引物的3'末端不应含有碱基修饰物,以免影响引物的扩增效率。
下面是几种常见的PCR引物设计方法:1.传统引物设计方法:传统引物设计方法主要是基于DNA序列的特点进行设计。
根据待扩增DNA片段的序列信息,可以选择合适的引物位置,并确保引物的长度、碱基组成和Tm值满足设计原则。
2.引物设计软件:引物设计软件是根据一系列预先设定的算法和规则,自动设计合适的引物。
常用的引物设计软件有Primer3、Primer-BLAST等。
这些软件可以根据用户输入的目标序列信息,自动生成合适的引物序列,并提供引物的Tm值、特异性等信息。
3.引物库:引物库是包含大量已设计好的引物序列的数据库。
研究人员可以直接从引物库中选择合适的引物序列,以节省时间和精力。
常用的引物库有NCBI的PrimerBank和UCSC的Primer Database。
4.引物修饰:5.引物交互作用:引物交互作用是指多对引物之间的交叉杂交,形成二聚体或多聚体结构。
通过设计引物之间的相互作用,可以提高PCR的特异性和扩增效率。
常用的引物交互作用方法有引物交叉互补法、引物竞争法等。
引物设计方法引物设计正向引物:(1)首先从目的蛋白的基因序列的开头选取近十五个左右的基因,然后放入Tm Calculator (/doc/d213587409.html,/webtools/tmc/)中检测Tm值,是否在65℃左右。
如果过低,则将取的基因数目增加,一直到Tm满足要求;如果过高,则减少所取的基因数目,满足Tm要求(bp 不能小于15bp,一般为15-30bp)。
另外,5’端不能改变,3’端必须以C或G结尾。
(2)在质粒载体上找到最佳的酶切位点,不要切掉质粒载体中的有用部分或者标签(质粒载体有分N-端与C-端),选择好酶切位点。
(3)使用ApE(软件,可下载),将目的蛋白的基因全部黏贴进去,检查其中所有的酶切位点(Enzyme——> Enzyme selector)。
如果我们选择的酶切位点在目的基因中存在,则此酶切位点不能使用(会导致基因产物无法获得);若选择的酶切位点不存在与目的基因中,则可以使用。
(4)质粒载体中选取的酶切位点,将其基因序列放到NEBcutter (/doc/d213587409.html,/NEBcutter2/)中进行更加精确的酶切位点显示(如:),再次确定。
(5)最后确定连接到引物上的酶切位点序列需要充分考虑ORF 的要求(即三个基因确定一个蛋白质,错位的话会导致目的产物错误),从ATG开始,不能发生错位。
(例如:CTT GCG GCC GC G AAT TC A中)TCA 为一组,而酶切位点到TC结束,必须将A加上。
然后从酶切位点开始,如上图即从AATTC中的T开始往左数16bp的基因(至少15bp,15-18bp)。
然后将其连接到(1)中满足Tm的基因的5’端。
(如:CTT GCG GCC GC G AAT TC A atggcg gatgaagcca c)附:如果引物中酶切位点最后为ATG 而与其相连的目的蛋白的基因开始也是ATG 则可以删除一组,留一组即可。
PCR引物设计方法综述PCR(聚合酶链反应)是一种常用的分子生物学技术,可以在体外快速扩增DNA序列,具有高度的特异性和敏感性。
PCR的成功与否很大程度上取决于引物的设计。
引物是PCR反应中的关键部分,其设计的合理性直接影响PCR扩增的效果。
因此,正确选择和设计引物对于PCR的成功至关重要。
PCR引物设计的原则主要包括以下几点:1. 引物长度:引物的长度通常在18-30个碱基对之间。
过短的引物可能导致非特异性扩增,而过长的引物则可能影响PCR反应的效率。
2. 引物序列:引物的序列应该与目标序列的两端互相衔接,保证引物与模板DNA的互补性。
同时,引物的序列应该防止高度重复的区域和易产生二级结构的序列。
3. 引物间的互补性:引物之间的互补性会导致引物二聚体的形成,从而影响PCR的特异性。
因此,在设计引物时需要防止引物之间的互补性。
在PCR引物设计中,有多种方法和工具可以援助探究人员选择和设计合适的引物。
1. 序列比对与分析:起首,我们需要从已知的目标DNA 序列中选择一段适当的区域进行扩增。
通过序列比对和分析工具,如BLAST、EMBOSS等,我们可以找到与目标序列高度一致的区域,然后依据该区域设计引物。
2. 引物设计工具:许多在线工具可用于设计引物,如Primer3、Beacon Designer等。
这些工具可以依据给定的目标序列信息自动生成一对适当的引物。
3. 引物碱基组成的计算:引物碱基组成的计算可以援助评估引物的特异性和二级结构问题。
通常,引物的GC含量应在40%-60%之间。
4. 引物特异性的验证:引物特异性的验证是PCR引物设计过程中的重要一步。
可以通过引物在目标序列以及可能存在的非特异性目标上扩增的试验来验证引物的特异性。
PCR引物设计对于探究人员开展PCR试验和相关探究具有重要意义。
合理设计的引物可以保证PCR反应的特异性、敏感性和扩增效率。
虽然目前有许多方法和工具可用于引物设计,但探究人员依旧需要依据详尽试验需求和目标序列的特点灵活选择和设计引物。
引物设计的原理和程序引物设计是在分子生物学领域中非常重要的一项技术,用于在DNA或RNA序列中选择特定的区域进行放大、克隆或检测。
引物设计的目标是选择具有高度特异性和高效性的引物,以确保所需的目标序列可以准确地被扩增或检测到。
以下是引物设计的原理和程序的详细说明。
1.特异性:引物应该在目标区域具有高度特异性,即只与目标序列配对,并排除与非目标序列的配对。
这可以通过确保引物序列在目标区域的起始位置和长度与目标序列相匹配来实现。
2.合成效率:引物应该具有高合成效率,以确保引物在PCR或其他实验过程中可以被充分放大或扩增。
合成效率可以通过一些计算方法如GC 含量、引物长度和翻转重复等特征进行评估。
3.避免互补配对:引物设计时应避免两个引物之间或引物与DNA模板之间形成互补配对。
互补配对可能会导致引物之间的二聚体形成或降低引物与目标区域的结合能力。
引物设计通常分为两个主要步骤:目标序列选择和引物设计。
1.目标序列选择:选择目标序列是引物设计的第一步。
目标序列通常是想要扩增或检测的DNA或RNA片段。
这可以是已知序列的基因、intergenic区域、病毒或细菌的片段等。
目标序列的选择需要考虑研究的目的以及实验的具体要求。
2.引物设计:引物设计是根据目标序列选择适当的引物。
引物设计可以通过多种计算机程序或在线工具来实现。
a.引物长度选择:引物长度通常在18到30个碱基对之间,具体长度的选择取决于目标序列的特点和实验需求。
较长的引物可以提供更好的特异性,但可能会降低扩增效率。
b.GC含量计算:GC含量是引物设计中的一个重要参数,通常在40%到60%之间。
GC含量高的引物可以提供更好的热稳定性和特异性,但过高或过低的GC含量都可能影响引物的性能。
c.特异性评估:引物的特异性可以通过与非目标序列进行比对来评估。
在引物设计过程中,首先应排除与非目标序列存在高度相似度的区域,然后检查目标序列中引物能否与其他非目标序列配对。
引物设计正向引物:(1)首先从目的蛋白的基因序列的开头选取近十五个左右的基因,然后放入Tm Calculator (/webtools/tmc/)中检测Tm值,是否在65℃左右。
如果过低,则将取的基因数目增加,一直到Tm满足要求;如果过高,则减少所取的基因数目,满足Tm要求(bp 不能小于15bp,一般为15-30bp)。
另外,5’端不能改变,3’端必须以C或G结尾。
(2)在质粒载体上找到最佳的酶切位点,不要切掉质粒载体中的有用部分或者标签(质粒载体有分N-端与C-端),选择好酶切位点。
(3)使用ApE(软件,可下载),将目的蛋白的基因全部黏贴进去,检查其中所有的酶切位点(Enzyme——> Enzyme selector)。
如果我们选择的酶切位点在目的基因中存在,则此酶切位点不能使用(会导致基因产物无法获得);若选择的酶切位点不存在与目的基因中,则可以使用。
(4)质粒载体中选取的酶切位点,将其基因序列放到NEBcutter (/NEBcutter2/)中进行更加精确的酶切位点显示(如:),再次确定。
(5)最后确定连接到引物上的酶切位点序列需要充分考虑ORF的要求(即三个基因确定一个蛋白质,错位的话会导致目的产物错误),从ATG开始,不能发生错位。
(例如:CTT GCG GCC GC G AAT TC A中)TCA 为一组,而酶切位点到TC结束,必须将A加上。
然后从酶切位点开始,如上图即从AATTC中的T开始往左数16bp的基因(至少15bp,15-18bp)。
然后将其连接到(1)中满足Tm的基因的5’端。
(如:CTT GCG GCC GC G AAT TC A atggcg gatgaagcca c)附:如果引物中酶切位点最后为ATG 而与其相连的目的蛋白的基因开始也是ATG 则可以删除一组,留一组即可。
例:Nde1 Ufm 1 FOR CGCGCGGCAGC CATATG(atg)tcgaaggtttcctttaagatcac反向引物:步骤与正向引物相似,不过(1)从目的蛋白的基因的3’-端开始往前取,取到满足Tm值的。
Primer 5.0搜索引物:1.Primer Length我常设置在18-30bp,短了特异性不好,长了没有必要。
当然有特殊要求的除外,如加个酶切位点什么的。
2.PCR Product size最好是100-500bp之间,小于100bp的PCR产物琼脂糖凝胶电泳出来,条带很模糊,不好看。
至于上限倒也不必要求苛刻。
3.Search parameters还是选Manual吧,Search stringency应选High,GC含量一般是40-60%。
其它参数默认就可以了。
4.搜索出来的引物,按Rating排序,逐个送Oligo软件里评估。
当然,搜索出的引物,其扩增产物很短,你可以不选择它,或是引物3端≥2个A或T,或引物内部连续的G或C太多,或引物3端≥2个G或C,这样的引物应作为次选,没得选了就选它。
对于这样的引物,如果其它各项指标还可以,我喜欢在引物末端去掉一个不满意的或加上一个碱基,看看引物的评估参数有没有变好点。
Oligo 6.0评估引物:1.在analyze里,Duplex Formation不管是上游引物、下游引物还是上下游引物之间,The most stable 3’-Dimer绝对值应小于4.5kcal/mol, The most stable Dimer overall绝对值一般应小于多少kcal/mol跟PCR退火温度有关,我几次实验感觉在PCR退火温度在65°的时候,The most stable Dimer overall 6.7kcal/mol没有问题。
2.Hairpin Formation根据黄金法则3.False priming sites: Primer的priming efficiency应该是错配地方的4倍左右,更多当然更好。
4.在PCR栏,个人感觉其所显示的optimal annealing temperature数值值得参考。
在PCR摸索条件的时候,退火温度为其数值加减2的范围就可以了。
5.Internal stability很重要:我们希望引物的内部稳定性是中间高、两边低的弧形,最起码保证3端不要过于稳定。
下图1引物3端过于稳定,很容易导致不适当扩增。
△G参照黄金法则,这其实很好理解:把一滴水放到大海里,这滴水就会不停的扩散分布,扩散的越厉害越稳定,所以△G绝对值越大结构越稳定。
最后说一句,敢于尝试就会成功。
第二贴--科室工作很多,小医生了,没有办法,所以肯怕不能满足很多战友的要求(qq聊或帮助设计),在此表示抱歉。
就楼上的问题我试着回答一下,不一定正确,供参考吧。
--1、两个评价系统不一样,个人感觉oligo评价引物好点,primer出来的引物,我一般按效率排序,再结合退火温度和引物长度,选择引物到oligo测试。
这是初步的选择,其实引物到了oligo里,退火温度也不一样。
--2、3端的二聚体应该避免,这个要看你的退火温度决定,一个50°的退火温度肯定和65°对二聚体的影响不一样了,一般来讲尽量控制在-4.5kcal/mol以下(个人观点,很多东西真得还是需要自己摸索)。
--3、个人感觉3端有A无A影响不大,3端有T的没有经验。
有T是不是一定不行,个人感觉不见得。
软件是评估,法则也不是没有例外,不是1+1=2那么确定。
--4、错配和二聚体谁轻谁重,个人觉得“到致命的程度”谁都重要,我也说不好。
我设计的时候,尽量两个都不得罪。
--5、GC含量并非不重要,它直接影响引物各端稳定性,3端来两个G或C,稳定性就上去了,粘在模板上很牢。
所以我设计的时候,尽量避免这样的情况出现。
谈一下我学这个引物设计的过程吧:入门--前面我说过是我的大师兄把我领进门的,之后我就自己捣鼓。
一开始严格按照引物设计的黄金法则来,primer搜出来的引物oligo评估,我就这样一条引物一条引物地反复评估,不厌其烦,到处查阅资料,一做就是几个小时,幸运的是捣鼓出来一两条符合标准的引物。
提高--有了成功的经验后,就继续做,当时我们实验室要定很多引物,我就有了练手的素材,这也很重要--什么东西不是练出来?!设计多了,问题也多了。
有些引物按黄金法则来,行不通了,怎么办?1、我就把引物3端或5端延长或缩短一个或几个碱基,再反复评估2、有些引物还不行,我就把5端突变一个碱基,再反复评估3、再不行,我就放宽条件。
放宽到什么样可以,不好说,自己摸索。
有的一对引物,其中一条错配很厉害,而另一条很可以,在没有其它选择的情况下为什么不可以。
有的人担心,这不一定跑的出来啊。
对自己要有信心啊,原因有二:1.自己的引物谁有功夫帮你设计?所以啊,当时我就被“赶鸭子上架了”,反反复复搜索评估的基础上,出来的应该是目前最好的;2、还有,我经常看国外文章的引物,有的评估起来表面上差的要命,人家照用,结果还不是出来了。
我就分析这样的引物,得出经验,胆子也大了。
当然失败肯定是有一两次的。
当然,有人说我就设计那么一条引物,输不起。
那只好说Sorry了!总之,我的经验就是:立即上手,遇到问题再解决问题,不厌其烦,不就是个软件嘛,还征服不了?!在NCBI上搜索到该基因,找到该基因的mRNA,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。
打开Primer Premier5,点击File-New-DNA sequence, 出现输入序列窗口,Copy目的序列在输入框内(选择As),此窗口内,序列也可以直接翻译成蛋白。
点击Primer,进入引物窗口。
此窗口可以链接到“引物搜索”、“引物编辑”以及“搜索结果”选项,点击Search按钮,进入引物搜索框,选择“PCR primers”,“Pairs”,设定搜索区域和引物长度和产物长度。
在Search Parameters里面,可以设定相应参数。
一般若无特殊需要,参数选择默认即可,但产物长度可以适当变化,因为100~200bp的产物电泳跑得较散,所以可以选择300~500bp。
点击OK,软件即开始自动搜索引物,搜索完成后,会自动跳出结果窗口,搜索结果默认按照评分(Rating)排序,点击其中任一个搜索结果,可以在“引物窗口”中,显示出该引物的综合情况,包括上游引物和下游引物的序列和位置,引物的各种信息等。
对于引物的序列,可以简单查看一下,避免出现下列情况:3’端不要以A结尾,最好是G或者C,T也可以;3’不要出现连续的3个碱基相连的情况,比如GGG或CCC,否则容易引起错配。
此窗口中需要着重查看的包括:Tm应该在55~70度之间,GC%应该在45%~55%间,上游引物和下游引物的Tm值最好不要相差太多,大概在2度以下较好。
该窗口的最下面列出了两条引物的二级结构信息,包括,发卡,二聚体,引物间交叉二聚体和错误引发位置。
若按钮显示为红色,表示存在该二级结构,点击该红色按钮,即可看到相应二级结构位置图示。
最理想的引物,应该都不存在这些二级结构,即这几个按钮都显示为“None”为好。
但有时很难找到各个条件都满足的引物,所以要求可以适当放宽,比如引物存在错配的话,可以就具体情况考察该错配的效率如何,是否会明显影响产物。
对于引物具体详细的评价需要借助于Oligo来完成,Oligo自身虽然带有引物搜索功能,但其搜索出的引物质量感觉不如Primer5。
在Primer5窗口中,若觉得某一对引物合适,可以在搜索结果窗口中,点击该引物,然后在菜单栏,选择File-Print-Current pair,使用PDF虚拟打印机,即可转换为Pdf文档,里面有该引物的详细信息。
引物评价在Oligo软件界面,File菜单下,选择Open,定位到目的cDNA序列(在primer中,该序列已经被保存为Seq文件),会跳出来两个窗口,分别为Internal Stability(Delta G)窗口和Tm 窗口。
在Tm窗口中,点击最左下角的按钮,会出来引物定位对话框,输入候选的上游引物序列位置(Primer5已经给出)即可,而引物长度可以通过点击Change-Current oligo length 来改变。
定位后,点击Tm窗口的Upper按钮,确定上游引物,同样方法定位下游引物位置,点击Lower按钮,确定下游引物。
引物确定后,即可以充分利用Analyze菜单中各种强大的引物分析功能了。
Analyze中,第一项为Key info,点击Selected primers,会给出两条引物的概括性信息,其中包括引物的Tm值,此值Oligo是采用nearest neighbor method计算,会比Primer5中引物的Tm值略高,此窗口中还给出引物的Delta G和3’端的Delta G。
3’端的Delta G过高,会在错配位点形成双链结构并引起DNA聚合反应,因此此项绝对值应该小一些,最好不要超过9。
Analyze中第二项为Duplex Formation,即二聚体形成分析,可以选择上游引物或下游引物,分析上游引物间二聚体形成情况和下游引物间的二聚体情况,还可以选择Upper/Lower,即上下游引物之间的二聚体形成情况。
引物二聚体是影响PCR反应异常的重要因素,因此应该避免设计的引物存在二聚体,至少也要使设计的引物形成的二聚体是不稳定的,即其Delta G值应该偏低,一般不要使其超过4.5kcal/mol,结合碱基对不要超过3个。
Oligo此项的分析窗口中分别给出了3’端和整个引物的二聚体图示和Delta G值。
Analyze中第三项为Hairpin Formation,即发夹结构分析。
可以选择上游或者下游引物,同样,Delta G值不要超过4.5kcal/mol,碱基对不要超过3个。
Analyze中第四项为Composition and Tm,会给出上游引物、下游引物和产物的各个碱基的组成比例和Tm值。
上下游引物的GC%需要控制在40%~60%,而且上下游引物之间的GC%不要相差太大。
Tm值共有3个,分别采用三种方法计算出来,包括nearest neighbor method、%GC method和2(A+T)+4(G+C)method,最后一种应该是Primer5所采用的方法,Tm值可以控制在50~70度之间。
第五项为False Priming Sites,即错误引发位点,在Primer5中虽然也有False priming分析,但不如oligo详细,并且oligo会给我正确引发效率和错误引发效率,一般的原则要使错误引发效率在100以下,当然有时候正确位点的引发效率很高的话,比如达到400~500,错误引发效率超过100幅度若不大的话,也可以接受。
Analyze中,有参考价值的最后一项是“PCR”,在此窗口中,是基于此对引物的PCR反应Summary,并且给出了此反应的最佳退火温度,另外,提供了对于此对引物的简短评价。