着色性干皮病临床案例
- 格式:pptx
- 大小:78.70 KB
- 文档页数:3


日晒伤ꎬ随着日光照射时间累积ꎬ进行性出现增多的不规则雀斑样皮损ꎬ伴色素沉着㊁毛细血管扩张ꎬ皮肤干燥㊁粗糙ꎬ可出现日光性角化病ꎬ角化棘皮瘤甚至恶性肿瘤[2ꎬ3]ꎬ如基底细胞癌㊁鳞状细胞癌ꎬ少数情况下继发黑素瘤ꎮ据研究报道ꎬXP患者与普通人群相比ꎬ出现非黑色素细胞性皮肤癌的风险高达10ꎬ000倍ꎬ出现黑色素瘤风险是2000倍ꎮ这种增加的癌症风险导致XP患者诱发非黑素细胞性皮肤癌年龄缩短58岁ꎬ诱发黑色素瘤年龄缩短33岁[1]ꎮPTEN突变是紫外线辐射诱发皮肤变化的重要特征ꎬDNA测序研究显示ꎬPTEN突变率在XP黑色素瘤中高达53%ꎬ而在普通人群中突变率只有16%[4]ꎮ约75%XP患者可出现眼部损害ꎬ如畏光㊁流泪ꎬ结膜炎ꎬ睑球粘连ꎬ睑外翻以及眼睑肿瘤等ꎮ此外20%~30%患者有神经系统疾病和智力障碍ꎬ如综合征XP[5]ꎮ该报道患者是一个典型的XP病例ꎮ患者皮肤光敏感ꎬ色素沉着过度ꎬ面部基底细胞癌病史ꎬ符合XP的诊断ꎮ该病鉴别诊断包括:(1)Peutz-Jeughers综合征(Peutz-JeughersSyndromeꎬPJS)又称家族性黏膜皮肤色素沉着胃肠道息肉病ꎬ它呈常染色体显性遗传ꎬ与STK11基因突变有关ꎬ一般在出生时或出生后不久发生ꎬ表现为口腔㊁鼻等腔口部位色素斑点ꎬ常有腹痛㊁腹泻㊁便血等腹部症状ꎬ伴胃肠错构瘤样息肉且易恶变ꎻ(2)泛发性黑子:包括发疹性黑子病和多发性黑子综合征ꎮ前者发病迅速且较突然ꎬ短时间内出现大量黑子ꎬ后者常有家族史ꎬ属常染色体显性遗传病ꎬ表现为多发性黑子伴多种先天性缺陷ꎬ如心脏发育异常ꎬ肺动脉或主动脉狭窄等ꎻ(3)COFS综合征是一种常染色体隐性遗传性疾病ꎬ为科卡尼综合征(CS)中病情最为严重的一类ꎮ又称先天CS综合征ꎬ与三种DNA修复基因CBS㊁XPD和XPG突变有关ꎮ特征为小头畸形ꎬ先天性白内障ꎬ重度智障ꎬ面部异形ꎬ关节挛缩ꎮ治疗方面ꎬ尽量避免日晒ꎬ建议遮光剂保护皮肤ꎬ同时做好预防ꎬ对患者家族中出现的早期皮损给予及时保护ꎮ类维生素A的系统治疗有研究报道可以减少皮肤癌的发生[3]ꎮ早期切除癌前期恶性病变对于长期生存很重要ꎬ此外使用基因治疗和抗氧化剂来减少氧化损伤的检测疗法可能成为未来治疗的新选择[6]ꎮ参考文献[1]KarassMꎬNaguibMNꎬElawabdehNꎬetal.Xerodermapig ̄mentosa:threenewcaseswithanindepthreviewofthege ̄neticandclinicalcharacteristicsofthedisease[J].FetalPe ̄diatrPatholꎬ2015ꎬ34(2):120-127.[2]郑金锋ꎬ王丽ꎬ刘翠杰.幼儿着色性干皮病伴角化棘皮瘤1例[J].中国麻风皮肤病杂志ꎬ2014ꎬ30(7):432-433. [3]孙景英ꎬ于世荣ꎬ普雄明ꎬ等.着色性干皮病继发基底细胞癌一例[J].中国麻风皮肤病杂志ꎬ2016ꎬ32(3):175-176.[4]MasakiTꎬWangYꎬDiGiovannaJJꎬetal.HighfrequencyofpTENmutationsinneviandmelanomasfromxerodermapig ̄mentosumpatients[J].PigmentCellMelanomaResꎬ2014ꎬ7(3):454-64.[5]蒋毅ꎬ胡杏ꎬ陈明亮.着色性干皮病痴呆综合征1例[J].临床皮肤科杂志ꎬ2011ꎬ40(5):297-298.[6]DupuyAꎬSarasinA.DNAdamageandgenetherapyofxero ̄dermapigmentosumꎬahumanDNArepair-deficientdisease[J].MutatResꎬ2015ꎬ776:2-8.(收稿:2018-10-23㊀修回:2018-11-08)口服小剂量甲泼尼龙治愈儿童普秃一例孙勇虎㊀张福仁㊀㊀病例资料㊀患儿ꎬ女ꎬ6岁ꎮ因 头发缓慢脱落1年ꎬ全部脱落1月余 就诊ꎮ患者1年前无明显诱因开始头发稀疏脱落ꎬ逐渐加重ꎬ1月前头发和眉毛完全脱落ꎮ曾在当地使用胸腺肽等治疗ꎬ效果不佳ꎬ当地医院转诊至我院门诊就诊ꎮ体检:一般情况可ꎬ系统查体未及明显异常ꎮ皮肤科情况:头发及眉毛完全脱落(图1a)ꎮ实验室检查:血常规㊁肝肾功能无异常ꎮ作者单位:山东省皮肤病性病防治研究所ꎬ山东济南ꎬ250022通信作者:张福仁ꎬE-mail:zhangfuren@hotmail.com㊀㊀诊断及治疗:根据临床表现诊断为:普秃ꎮ给予小剂量甲泼尼龙8mg/d口服治疗ꎬ治疗2周后毛发虽有生长但不明显(图1b)ꎬ1个月后头发已开始稀疏生长ꎬ初起细软ꎬ后逐渐生长变硬ꎬ覆盖整个头皮(图1c)ꎮ治疗3个月后ꎬ毛发生长迅速ꎬ虽留有数个无毛发区ꎬ但毛发覆盖已超过80%头皮面积ꎬ眉毛已基本正常生长ꎬ甲泼尼龙片减量至4mg(图1d㊁1e)ꎮ治疗5个月后ꎬ头发逐渐覆盖无毛发区ꎬ部分较正常稍显稀疏(图1f)ꎮ治疗7个月ꎬ头发及眉毛已生长完全ꎬ停止服药(图1g)ꎮ停药5个月后门诊随访ꎬ头发仍正常生长ꎬ未见脱落(图1h)ꎮ停药1年后电话随访992中国麻风皮肤病杂志㊀2019年5月第35卷第5期㊀㊀1a:治疗前ꎻ1b:治疗2周ꎻ1c:治疗1个月ꎻ1d:治疗3个月(俯视)ꎻ1e:治疗3个月(平视)ꎻ1f:治疗5个月ꎻ1g:治疗7个月(停药)ꎻ1h:停药5个月后图1㊀患者治疗前后毛发生长临床图片毛发生长正常ꎮ治疗及随访过程中患儿生长发育情况未见异常ꎮ㊀㊀讨论㊀全秃是指头发的全部脱落ꎬ而普秃是指全身所有毛发的脱落ꎬ目前尚缺乏公认的可靠安全的系统性治疗方法ꎮ光化学疗法(PUVA)㊁外用封包强效糖皮质激素㊁系统应用糖皮质激素及免疫抑制剂(环孢素㊁甲氨蝶呤和硫唑嘌呤)等治疗斑秃㊁全秃及普秃均有报道ꎬ但效果不一ꎮ随着生物制剂的临床应用ꎬ利用TNF-α拮抗剂和JAK激酶拮抗剂来治疗秃发性疾病也多有报道[1]ꎮ2017年ꎬIbrahim等报道13例用JAK激酶抑制剂托法替尼治疗的斑秃及全秃病例ꎬ7例患者的毛发生长率在1~9个月的区间里生长了超过50%[2]ꎮ本例患者为6岁儿童ꎬ头发及眉毛无明显诱因短时间内全部脱落ꎬ应用小剂量甲泼尼龙口服后起效较快ꎬ头发及眉毛在3个月左右已完全恢复正常生长ꎬ7个月毛发覆盖全部头皮ꎬ停药5个月后门诊随访未见有新的头发脱落出现ꎮ目前已停药1年ꎬ电话随访未出现新的头发脱落ꎬ用药过程及结束后生长发育无明显异常ꎮ本病例提示对于儿童期普秃的治疗ꎬ口服小剂量甲泼尼龙安全有效ꎮ参考文献[1]KassiraSꎬKortaDZꎬChapmanLWꎬetal.Reviewoftreat ̄mentforalopeciatotalisandalopeciauniversalis[J].IntJDermatolꎬ2017ꎬ56(8):801-810.[2]IbrahimOꎬBayartCBꎬHoganSꎬetal.Treatmentofalopeciaareatawithtofacitinib[J].JAMADermatolꎬ2017ꎬ153(6):600-602.(收稿:2018-12-11㊀修回:2018-12-23)003ChinJLeprSkinDis.May2019ꎬVol.35ꎬNo.5。

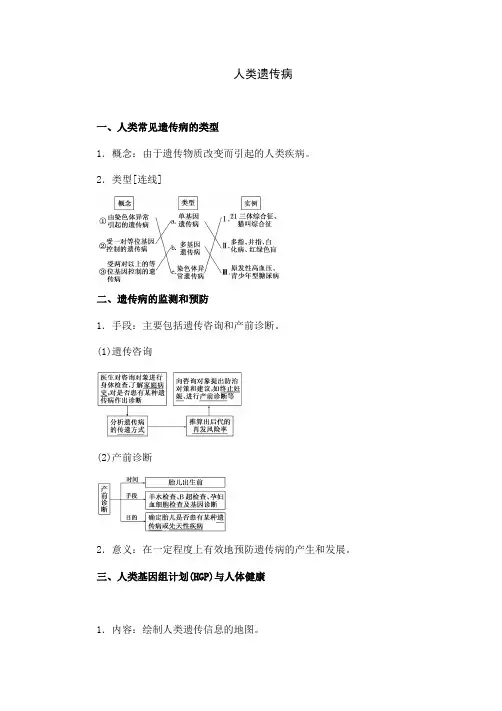
人类遗传病一、人类常见遗传病的类型1.概念:由于遗传物质改变而引起的人类疾病。
2.类型[连线]二、遗传病的监测和预防1.手段:主要包括遗传咨询和产前诊断。
(1)遗传咨询(2)产前诊断2.意义:在一定程度上有效地预防遗传病的产生和发展。
三、人类基因组计划(HGP)与人体健康1.内容:绘制人类遗传信息的地图。
2.目的:测定人类基因组的全部DNA序列,解读其中包含的遗传信息。
3.意义:认识人类基因的组成、结构、功能及其相互之间的关系,对人类疾病的诊治和预防具有重要意义。
4.结果:人类基因组由大约31.6亿个碱基对组成,已发现的基因约为2.0万~2.5万个。
重点聚焦1.人类常见的遗传病有哪些类型,病因是什么?2.如何监测和预防遗传病?3.人类基因组计划的目的和内容是什么?[共研探究]1.单基因遗传病有6 500多种,并且每年在以10~50种的速度递增,单基因遗传病已经对人类健康构成了较大的威胁,常见的有红绿色盲、血友病、白化病等。
与单基因遗传病相比,多基因遗传病不只由遗传因素决定,而是遗传因素与环境因素共同起作用。
根据上述资料,回答下列问题:(1)单基因遗传病是受一对等位基因控制的遗传病。
多基因遗传病受两对以上的等位基因控制。
(2)单基因遗传病和多基因遗传病说明:一种相对性状可以由一对或多对基因控制。
(3)单基因遗传病遵循孟德尔遗传定律,多基因遗传病和染色体异常遗传病不遵循孟德尔遗传定律。
(4)人类遗传病中单基因遗传病和多基因遗传病都是由致病基因引起的,染色体异常遗传病不是由致病基因引起的。
2.21三体综合征患者的体细胞中含有3条21号染色体,是由于亲本产生的异常配子发生受精作用导致的。
如图表示21三体综合征患者产生异常卵细胞的示意图(图中染色体代表21号染色体)(1)异常卵细胞的形成①减数第一次分裂后期同源染色体未分离,产生异常的次级卵母细胞,进而形成了含有两条21号染色体的异常卵细胞。
②减数第二次分裂后期姐妹染色单体分离形成的染色体未被拉向两极,产生了含有两条21号染色体的异常卵细胞。







第二节酶的高效催化和专一性的机制李新梅湖南大学生物学院第二节知识结构酶催化的专一性和高效性的机制一、什么是酶的活性中心二、为什么酶具有高效催化性?三、为什么酶催化具有专一性?一、什么是酶的活性中心李新梅湖南大学生物学院案例1:投弹手甲虫-昆虫界的炮兵⏹1、投弹手甲虫可以在瞬间喷射高温高压的刺激性液体–是一种高效的酶促反应⏹即:+2H ⏹(2)过氧化氢酶,酶学分类号为EC 1.11.1.6–存在于所有已知的动物的各个组织中,特别在肝脏中以高浓度存在–催化反应是2 H 2O 2→O 2+2H 2O+Q李新梅湖南大学生物学院李新梅湖南大学生物学院⏹投弹手甲虫腹部具有两套分开储存于腺体中的化学物–辣根过氧化物酶⏹过氧化氢分解产生氧和水,形成高温的水蒸李新梅湖南大学生物学院李新梅湖南大学生物学院3高效性小空间内两个相遇且碰撞发生反应李新梅湖南大学生物学院–酶的活性中心active site active site、活性中心结合部位催化部位底物的敏感键在此处断裂而形成新键,决定酶的高效性李新梅湖南大学生物学院羧心示意图3维持李新梅湖南大学生物学院李新梅李新梅湖南大学生物学院李新梅湖南大学生物学院AspHisSer胰凝乳蛋白酶的活性中心李新梅湖南大学生物学院⏹胰凝乳蛋白酶的活性中心李新梅湖南大学生物学院⏹(3)经过底物诱导后互补的三维空间结构–与底物互补的酶分子表面的一个三维的空间凹穴活性中心特殊的构象,在与底物接触时表现一定的柔性,底物分子诱导酶的活性中心发生变化形成互补构象X -Y学习小结生物分子⏹判断:酶活性中心是酶分子的一小部分。
(⏹下面关于酶的描述,哪一项不正确:⏹酶催化底物时将产生哪种效应––⏹三、酶为什么具有专一性?李新梅湖南大学生物学院⏹1.锁钥学说–1894 Fischer 提出–认为底物分子或底物分子的一部分象钥匙那样,专一地楔入到酶的活性中心部位–不能解释酶的逆反应–不能解释酶专一性中的所有现象李新梅湖南大学生物学院酶专一性的“锁钥学说”这种学说不能解释酶活性中心的结构与底物和产物结构都吻合的现象,也不能解释酶专一性中的所有现象李新梅湖南大学生物学院2.诱导契合学说获得认可⏹1958年,Koshland 提出诱导契合–酶表面并没有一种与底物互补的固定形状–当酶分子与底物分子接近时,酶蛋白受底物分子诱导,其构象发生有利于底物结合的变化,酶与底物在此基础上互补契合进行反应。
表皮色素增加性皮肤病中,色素异常表现为多种形式。
有的表现为黑素细胞的功能形态异常,如雀斑;有的表现为黑素细胞数量增加,如雀斑样痣。
而有些则仅为噬黑素细胞增加,如黄褐斑和炎症后色素沉着。
一般而言,由黑素细胞数量增加所致者激光治疗效果较好,而由于全身或局部代谢异常所致的黄褐斑、炎症后色素沉着等激光治疗效果不佳。
一、雀斑雀斑(freckles,ephelides)是常见的好发于中青年女性的色素沉着病。
【病因和发病机理】本病是常染色体显性遗传性疾病。
雀斑皮肤黑素细胞内的酪氨酸酶活性增加,在日光、X线、紫外线甚至日光灯照射后,产生大量的黑素,形成斑点状色素沉着。
【临床表现】雀斑通常在幼儿期至学龄儿童发病,青春期常可增多。
女性多于男性。
本病常发生在暴露部位,特别是面部,尤以鼻和颊最为常见,少见于手背、前臂、颈、肩部。
皮损为1 mm~4 mm,圆形、椭圆形或多角形,边缘不规则的淡褐色至深褐色斑点,表面可略有凹陷,境界清楚,不融合,分布可疏密不一,对称或不对称。
雀斑与日晒关系密切,其色素斑点的数目、大小、颜色取决于吸收阳光的量及个体对阳光的耐受性,夏季雀斑的数目多、形体大,为深褐色,冬季则相反。
不同人种斑点色素可有不同。
【组织病理】皮损黑素细胞数目没有增加,用多巴染色可见雀斑的黑素细胞密度较周围正常皮肤减少,但雀斑的黑素细胞较周围皮肤的黑素细胞大而且有更多更长的树状突,染色比正常皮肤深。
用电镜观察,雀斑的黑素细胞产生大量椭圆形全黑素化颗粒,类似于黑种人的黑素细胞,相邻正常皮肤的黑素颗粒小、量少,两者有明显的差异。
【诊断及鉴别诊断】根据本病发生在暴露部位,孤立而不融合的棕褐色小斑点,日晒后加重,Wood灯下皮损色素更加明显等特点,易于诊断。
主要与雀斑样痣、面正中雀斑样痣,早期着色干皮病及色素沉着一肠道息肉综合征区别。
1. 单纯雀斑样痣皮损颜色常较雀斑深,呈黑褐色至黑色,与日晒无关,无夏重冬轻的变化,可发生在任何部位,但集簇于一定范围。
stain[J].JAmAcadDermatolꎬ1987ꎬ16(2):326-331. [5]TebbeBꎬStavropoulosPGꎬKrasagakisKꎬetal.Cutaneousmastocytosisinadults.evaluationof14patientswithrespecttosystemicdiseasemanifestations[J].Dermatologyꎬ1998ꎬ197(2):101-108.[6]杨凡方ꎬ江平ꎬ陈柳青.持久性发疹性斑状毛细血管扩张1例[J].中国麻风皮肤病杂志ꎬ2015ꎬ31(5):303-304. [7]KoldeGꎬFroschPJꎬCzarnetzkiBM.ResponseofcutaneousmastcellstoPUVAinpatientswithurticariapigmentosa:histomorphometricꎬultrastructuralꎬandbiochemicalinvesti ̄gations[J].JInvestDermatolꎬ1984ꎬ83(3):175-178. [8]BartonJꎬLavkerRMꎬSchechterNMꎬetal.Treatmentofur ̄ticariapigmentosawithcorticosteroids[J].ArchDermatolꎬ2016ꎬ121(12):1516-1519.[9]CzarnyJꎬLangeMꎬNiedoszytkoMꎬetal.Transientim ̄provementofskinsymptomsinanadultpatientwithpediatric-onsetcutaneousmastocytosistreatedwithinterferonα[J].IntJDermatolꎬ2018ꎬ57(10):1237-1241.(收稿:2019-01-30㊀修回:2019-04-22)着色性干皮病一家系王白鹤1㊀周鹏军2㊀孙建方1㊀㊀临床资料㊀患者ꎬ女ꎬ37岁ꎮ主因 面部发生雀斑样皮损31年 就诊ꎮ患者6岁时面部开始出现黑色斑点ꎬ并逐渐增多ꎬ泛发至全身ꎬ以面颈部为著ꎬ皮疹日晒后加重ꎮ半年前无明显诱因面中部斑点基础上出现一处褐色增生物ꎬ约黄豆大ꎬ就诊于当地医院ꎬ给予手术切除并行组织病理检查诊断 基底细胞癌 ꎬ之后未见复发ꎬ但面部多发雀斑样皮疹一直未消退ꎬ现为进一步治疗来我院ꎬ患者自发病以来精神睡眠可ꎬ二便正常ꎬ体重未见明显变化ꎮ体格检查:一般情况较好ꎬ各系统检查未见明显异常ꎬ发育和智力正常ꎮ皮肤科检查:面部及躯干上部㊁双上肢皮肤干燥ꎬ可见泛发性雀斑样皮损ꎬ褐色针尖至粟粒大ꎬ数目多ꎬ部分融合成不规则色素斑ꎬ可见白色萎缩斑点㊁疣状角化及毛细血管扩张ꎮ左面部可见小片术后瘢痕(图1aꎬb)ꎮ适龄结婚ꎬ育有一儿一女ꎬ配偶体健ꎬ子女均有类似皮肤表现ꎮ患者父母非近亲结婚ꎬ其妹妹和兄长亦有相似皮肤病史ꎮ家系图谱见(图2)ꎮ临床诊断:着色性干皮病ꎮ治疗:嘱患者避免日晒ꎬ给予遮光剂保护皮肤ꎻ同时建议其家属详细体格检查ꎬ对出现的早期皮损给予及时保护和预防ꎮ㊀㊀讨论㊀着色性干皮病(xerodermapigmentosumꎬXP)是一种常染色体隐性遗传皮肤病ꎮ该病在1870年被Kaposi首先报道ꎬ它包括7个互补型(XP-A到G)和一个变异型(XP-V)ꎮ其中XPC在美国ꎬ欧洲和北非最常见ꎬ而XPA是日本最常见的类型[1]ꎮXPV由DNA聚合酶G突变引起ꎬ与其他XP类型相基金项目:中国医学科学院医学与健康科技创新工程(编号:CIFMS-2017-I2M-1-107)作者单位:1中国医学科学院皮肤病研究所ꎬ南京ꎬ2100422福建医科大学附属协和医院ꎬ福州ꎬ350000通信作者:孙建方ꎬE-mail:fangmin5758@aliyun.com图1㊀aꎬb面部及躯干上部㊁双上肢皮肤干燥ꎬ可见泛发雀斑样皮损ꎬ褐色针尖至粟粒大ꎬ数目多ꎬ部分融合成不规则色素斑ꎬ可见白色萎缩斑点㊁疣状角化及毛细血管扩张ꎬ左面部可见小片术后瘢痕图2㊀着色性干皮病家系图比ꎬXPV患者的长期生存率可能增高ꎮ㊀㊀XP患者皮肤对日光照射高度敏感ꎮ多数患者因为缺乏核酸内切酶ꎬ无法修复紫外线照射后皮肤细胞DNA的损伤ꎬ常引发抑癌基因突变ꎬ因而患者易出现光损伤相关疾病和皮肤恶性肿瘤ꎮ此外ꎬ20%患者因缺乏DNA聚合酶无法完整修复DNA损伤而致病ꎬ如XPVꎮXP通常早年发病ꎬ起始年龄在6个月~3岁ꎬ病变局限于曝光部位ꎬ如面颈部㊁上肢伸侧等ꎬ初起轻微892ChinJLeprSkinDis.May2019ꎬVol.35ꎬNo.5日晒伤ꎬ随着日光照射时间累积ꎬ进行性出现增多的不规则雀斑样皮损ꎬ伴色素沉着㊁毛细血管扩张ꎬ皮肤干燥㊁粗糙ꎬ可出现日光性角化病ꎬ角化棘皮瘤甚至恶性肿瘤[2ꎬ3]ꎬ如基底细胞癌㊁鳞状细胞癌ꎬ少数情况下继发黑素瘤ꎮ据研究报道ꎬXP患者与普通人群相比ꎬ出现非黑色素细胞性皮肤癌的风险高达10ꎬ000倍ꎬ出现黑色素瘤风险是2000倍ꎮ这种增加的癌症风险导致XP患者诱发非黑素细胞性皮肤癌年龄缩短58岁ꎬ诱发黑色素瘤年龄缩短33岁[1]ꎮPTEN突变是紫外线辐射诱发皮肤变化的重要特征ꎬDNA测序研究显示ꎬPTEN突变率在XP黑色素瘤中高达53%ꎬ而在普通人群中突变率只有16%[4]ꎮ约75%XP患者可出现眼部损害ꎬ如畏光㊁流泪ꎬ结膜炎ꎬ睑球粘连ꎬ睑外翻以及眼睑肿瘤等ꎮ此外20%~30%患者有神经系统疾病和智力障碍ꎬ如综合征XP[5]ꎮ该报道患者是一个典型的XP病例ꎮ患者皮肤光敏感ꎬ色素沉着过度ꎬ面部基底细胞癌病史ꎬ符合XP的诊断ꎮ该病鉴别诊断包括:(1)Peutz-Jeughers综合征(Peutz-JeughersSyndromeꎬPJS)又称家族性黏膜皮肤色素沉着胃肠道息肉病ꎬ它呈常染色体显性遗传ꎬ与STK11基因突变有关ꎬ一般在出生时或出生后不久发生ꎬ表现为口腔㊁鼻等腔口部位色素斑点ꎬ常有腹痛㊁腹泻㊁便血等腹部症状ꎬ伴胃肠错构瘤样息肉且易恶变ꎻ(2)泛发性黑子:包括发疹性黑子病和多发性黑子综合征ꎮ前者发病迅速且较突然ꎬ短时间内出现大量黑子ꎬ后者常有家族史ꎬ属常染色体显性遗传病ꎬ表现为多发性黑子伴多种先天性缺陷ꎬ如心脏发育异常ꎬ肺动脉或主动脉狭窄等ꎻ(3)COFS综合征是一种常染色体隐性遗传性疾病ꎬ为科卡尼综合征(CS)中病情最为严重的一类ꎮ又称先天CS综合征ꎬ与三种DNA修复基因CBS㊁XPD和XPG突变有关ꎮ特征为小头畸形ꎬ先天性白内障ꎬ重度智障ꎬ面部异形ꎬ关节挛缩ꎮ治疗方面ꎬ尽量避免日晒ꎬ建议遮光剂保护皮肤ꎬ同时做好预防ꎬ对患者家族中出现的早期皮损给予及时保护ꎮ类维生素A的系统治疗有研究报道可以减少皮肤癌的发生[3]ꎮ早期切除癌前期恶性病变对于长期生存很重要ꎬ此外使用基因治疗和抗氧化剂来减少氧化损伤的检测疗法可能成为未来治疗的新选择[6]ꎮ参考文献[1]KarassMꎬNaguibMNꎬElawabdehNꎬetal.Xerodermapig ̄mentosa:threenewcaseswithanindepthreviewofthege ̄neticandclinicalcharacteristicsofthedisease[J].FetalPe ̄diatrPatholꎬ2015ꎬ34(2):120-127.[2]郑金锋ꎬ王丽ꎬ刘翠杰.幼儿着色性干皮病伴角化棘皮瘤1例[J].中国麻风皮肤病杂志ꎬ2014ꎬ30(7):432-433. [3]孙景英ꎬ于世荣ꎬ普雄明ꎬ等.着色性干皮病继发基底细胞癌一例[J].中国麻风皮肤病杂志ꎬ2016ꎬ32(3):175-176.[4]MasakiTꎬWangYꎬDiGiovannaJJꎬetal.HighfrequencyofpTENmutationsinneviandmelanomasfromxerodermapig ̄mentosumpatients[J].PigmentCellMelanomaResꎬ2014ꎬ7(3):454-64.[5]蒋毅ꎬ胡杏ꎬ陈明亮.着色性干皮病痴呆综合征1例[J].临床皮肤科杂志ꎬ2011ꎬ40(5):297-298.[6]DupuyAꎬSarasinA.DNAdamageandgenetherapyofxero ̄dermapigmentosumꎬahumanDNArepair-deficientdisease[J].MutatResꎬ2015ꎬ776:2-8.(收稿:2018-10-23㊀修回:2018-11-08)口服小剂量甲泼尼龙治愈儿童普秃一例孙勇虎㊀张福仁㊀㊀病例资料㊀患儿ꎬ女ꎬ6岁ꎮ因 头发缓慢脱落1年ꎬ全部脱落1月余 就诊ꎮ患者1年前无明显诱因开始头发稀疏脱落ꎬ逐渐加重ꎬ1月前头发和眉毛完全脱落ꎮ曾在当地使用胸腺肽等治疗ꎬ效果不佳ꎬ当地医院转诊至我院门诊就诊ꎮ体检:一般情况可ꎬ系统查体未及明显异常ꎮ皮肤科情况:头发及眉毛完全脱落(图1a)ꎮ实验室检查:血常规㊁肝肾功能无异常ꎮ作者单位:山东省皮肤病性病防治研究所ꎬ山东济南ꎬ250022通信作者:张福仁ꎬE-mail:zhangfuren@hotmail.com㊀㊀诊断及治疗:根据临床表现诊断为:普秃ꎮ给予小剂量甲泼尼龙8mg/d口服治疗ꎬ治疗2周后毛发虽有生长但不明显(图1b)ꎬ1个月后头发已开始稀疏生长ꎬ初起细软ꎬ后逐渐生长变硬ꎬ覆盖整个头皮(图1c)ꎮ治疗3个月后ꎬ毛发生长迅速ꎬ虽留有数个无毛发区ꎬ但毛发覆盖已超过80%头皮面积ꎬ眉毛已基本正常生长ꎬ甲泼尼龙片减量至4mg(图1d㊁1e)ꎮ治疗5个月后ꎬ头发逐渐覆盖无毛发区ꎬ部分较正常稍显稀疏(图1f)ꎮ治疗7个月ꎬ头发及眉毛已生长完全ꎬ停止服药(图1g)ꎮ停药5个月后门诊随访ꎬ头发仍正常生长ꎬ未见脱落(图1h)ꎮ停药1年后电话随访992中国麻风皮肤病杂志㊀2019年5月第35卷第5期。
着色性干皮病一家系的XP基因突变检测目的检测1个着色性干皮病家系的XPA和XPC基因突变。
方法收集1个着色性干皮病家系的临床资料,采集先证者及其家庭成员的外周血标本,提取外周血白细胞中的DNA,采用PCR反应扩增XPA和XPC所有外显子编码区及其侧翼序列,通过对PCR反应产物直接测序进行序列分析。
结果该着色性干皮病家系未发现XPA和XPC的致病性基因突变。
结论尽管XPA和XPC是国内比较常见的着色性干皮病亚型,但该家系可能属于其他的亚型。
标签:着色性干皮病;突变;XPA ;XPC【Abstract】Objectives To identify pathogenic mutation of the XPA and XPC gene in one familial cases of xeroderma pigmentosum.MethodsWe collected blood samples and clinical data from one familial cases of xeroderma pigmentosum. Genomic DNA was extracted from peripheral blood. All the coding exons and exon-intron flanking sequences of XPA and XPC gene were amplified by polymerase chain reaction and products were analyzed by direct sequencing.ResultsNo XPA and XPC pathogenic mutation was found in this family with xeroderma pigmentosum.ConclusionsAlthough XPA and XPC is a more common subtypes with xeroderma pigmentosum in the domestic, but this family may be other subtypes.【key words 】Xeroderma Pigmentosum / XPA / XPC / mutation着色性干皮病(Xeroderma Pigmentosum,XP)是一种极其少见的常染色体隐性遗传性损容性皮肤病,严重危害患者身心健康。
写在课前的话正常皮肤的表面脂膜、角质层、黑素均对光线有一定的防御作用,特别是黑素的保护作用更大,是决定皮肤颜色的主要色素。
一般将色素性皮肤病分为色素增加如黄褐斑、雀斑和色素减退如白癜风两大类。
大多数色素障碍性疾病对于婴幼儿和儿童仅影响美观,但某些皮损可以为多系统疾病的诊断提供线索。
一、概述正常皮肤可以呈现出红、黄、棕及黑色。
影响肤色的因素包括血红蛋白(氧和状态或还原状态)、黑素(起决定性作用)、类胡萝卜素。
黑素在黑素细胞中特殊的细胞器-黑素小体中合成,然后转运至周围角质形成细胞中。
肤色有种族和部位差异,此与黑素小体的数目、大小、形态、分布和降解方式的不同有关。
表皮黑素细胞来源于神经嵴,在胚胎早期由神经嵴向表皮移行并分化。
主要分布于表皮基底层、毛囊、真皮、黏膜上皮、内耳、软脑膜和视网膜。
多数黑素向上随着表皮生长移行到角质层脱落,排除体外。
少数黑素转移到真皮,被嗜色素细胞吞噬,运送到血循,经肾脏排除。
色素性皮肤病的发病机制主要包括黑素母细胞移行和分化异常、黑素细胞数目增多或减少以及黑素合成或转运异常。
色素障碍性疾病在临床上可根据其色素是增加或减少和其分布是局限或泛发来进行鉴别。
色素减退性皮肤病包括局限性和弥漫性白斑,每种白斑又有较多的分类,作为临床医务工作者一定要掌握每种疾病的病变特点,熟知相关的鉴别诊断要点,从而做出正确的诊治。
二、色素减退性皮肤病(Hypopigementation)(一)局限性白斑(Localizedhypopigmentation)1.先天性局限性白斑包括无色素性痣、Ito色素减退症、结节性硬化症(叶状白斑)、贫血痣、斑驳病等。
以下不属于先天性局限性白斑的为()A.无色素性痣B.Ito色素减退症C.贫血痣D.白癜风正确答案:D解析:先天性局限性白斑包括无色素性痣、Ito色素减退症、结节性硬化症(叶状白斑)、贫血痣、斑驳病等。
白癜风为一种常见的后天性局部色素脱失性疾病。
(1)无色素性痣(NevusDepigmentosus,或AchromicNevus)无色素性痣是一种先天性非进行性色素减退斑,临床特征为白斑生后或生后不久出现,其分布及相对大小终身不变。