色谱分析第六章毛细柱气相色谱法
- 格式:doc
- 大小:88.50 KB
- 文档页数:9


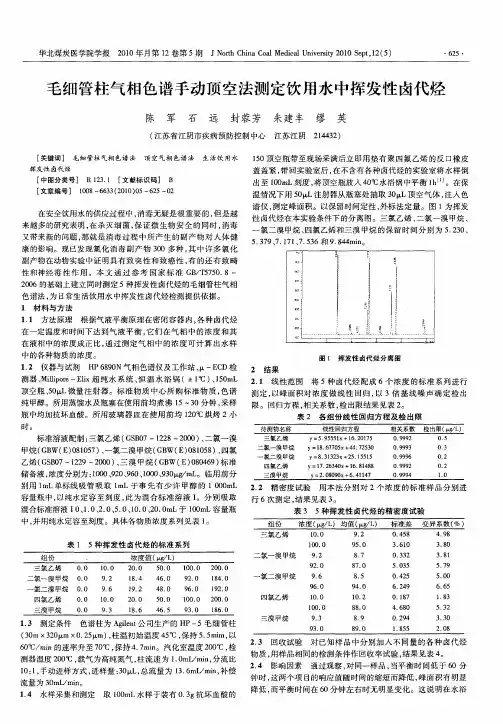



第32卷第3期 2018年9月干旱环境监测Arid Environmental MonitoringVol. 32 No.3See-• 2018毛细管柱气相色谱法测定有组织废气中氯乙烯施玉格,刘喜(新疆维吾尔自治区环境监测总站,新疆乌鲁木齐830011)摘要:利用毛细管柱对有组织废气中氯乙烯进行分离,火焰离子化检测器(F ID)进行检测,建立了有组织废气中氯乙烯的毛细管柱气相色谱测定法。
该方法检出限为0.08 mg/m3,空白样品加标回收率为98.6%~99.9%,变异系数为0.96%~1.24%。
实验结果表明,该方法准确可靠、灵敏度好、分析速度快、操作简便,适用于有组织废气中氯乙烯的测定。
关键词:毛细管柱;气相色谱法;有组织废气;氯乙烯中图分类号:X830.2 文献标识码:B 文章编号:1007 -1504(2018)03-0106-03Determination of Vinyl Chloride in Organized Waste Gas by Gas Chromatography witli Capillary ColumnSHI Yu - /,LIU X i( Xinjiang Environmental Monitoring Centre,Xinjian/ Urumqi 830011,China)Abstract :A m ethod was developed for determination of vinyl chloride in organized waste /as by /as chromatography with FIDand capillary column. The detection limit of the method was 0. 08 mg/m3. The recovery rate of the b lank sampl 99. 9% , and the coefficient of variation is 0. 96% to 1.24% . The results show that the metho tive, fast and easy to operate. It is suitable for the determination of vinyl chloride in Key words:capillary column;/as chromatography;organized waste /a s;vinyl chloride氯乙烯是一种重要的化工原料,工业上主要 用于生产聚氯乙烯。

毛细管柱气相色谱法测定六六六、滴滴涕作者:赵微来源:《科技风》2018年第08期摘要:本文利用ECD检测器、WondaCap5毛细管色谱柱对六六六、滴滴涕进行分离研究,并建立了毛细管柱气相色谱法测定六六六、滴滴涕的方法。
确立了本检测方法实验条件,实验结果表明在采用恒温条件时几种物质的分离效果不理想,但采用程序升温时几种物质分离效果良好,采用分流进样方式效果明显优于不分流进样方式。
8种有机氯在0.05mg/L~1mg/L 之间线性关系良好,相关系数均在0.999以上,相对标准偏差均(RSD%)关键词:气相色谱法;六六六;滴滴涕1绪论六六六、滴滴涕是以苯为原料的有机氯农药,其物理化学性质稳定,不易分解,且难溶于水[1]。
也是一种对环境构成严重威胁的人工合成化合物,具有致癌、致畸、致突变效应,并具有神经毒性和内分泌干扰特性[2]。
因此,建立灵敏、快速、高效有机氯农药分析方法,对人类健康及资源保护具有重要意义。
目前测定六六六、滴滴涕的方法较多,如酶联免疫法、光学免疫法、气相色谱法、气质联用法等[3],其中气相色谱法因具有分离效果好、分析速度快、灵敏及样品用量少等一系列特点而被广泛应用[4]。
本实验采用适当升温条件与分流进样可以在较短的时间内将8种有机氯完全分离,方法简便、快速。
2实验部分2.1仪器和试剂仪器:岛津GC2014C气相色谱仪,ECD检测器,WondaCap5毛细管色谱柱(30m×0.25mm×0.32um),气相色谱工作站为GCSolution,微量注射器。
试剂:石油醚中8种有机氯农药混合溶液标准物质(证书编号05991701、北京海岸鸿蒙标准物质技术有限责任公司)、实验过程中使用的溶剂为石油醚(国药集团化学试剂有限公司),使用之前需要用全玻璃蒸馏装置蒸馏,用气相色谱仪测定无干扰峰后使用。
2.2实验方法将石油醚中8种有机氯农药混合溶液标准物质分别稀释为0.05mg/L、0.1mg/L、0.2mg/L、0.5mg/L、1.0mg/L系列标准溶液。

现代⾊谱分析试题第⼀章⾊谱法概论1. 综述各种⾊谱法的特点及其应⽤。
2. 简要说明⽓相⾊谱法(GLC、GSC)、⾼效液相⾊谱法(HPLC的各类⽅法)特点及其应⽤范围?3. 试⽐较⾊谱法(GC、HPLC)之间的异同?第⼆章⾊谱基本理论例1 采⽤3M⾊谱柱对A、B⼆组分进⾏分离,此时测得⾮滞留组分的t M值为0.9min ,A组分的保留时间(t R(A))为15.1min,B组分的t R为18.0min,要使⼆组分达到基线分离(R=1.5),问最短柱长应选择多少⽶(设B组分的峰宽为1.1 min)?解⽅法(1):由已知条件,得n B=16(18.0/1.1)2=4284r i,B=18.0-0.9/15.1-0.9=1.20K/B=18.0-0.9/0.9=19则因为所以故⽅法(2):同⽅法(1)得n B=4284;r i,B=1.20;K/B=19所以则n B (R=1.5)=16 (1.5)2(1.2/0.2)2[(1+19)/19]2=1425故L=n (R=1.5)H=14250.07=99.8cm1m1.A、B ⼆组分的分配系数之⽐为0.912,要保证⼆者的分离度达到1.20,柱长因应选择多少⽶?设有效塔板⾼度为0.95mm.。
2.有⼀液相⾊谱柱长25cm,流动相速度为0.5ml/min,流动相体积为0.45ml,固定相体积为0.25ml,现测得萘、蒽、菲、芘四组分(以A、B、C、D)的保留值及峰宽如表3-1。
根据已知条件试计算出:(1)各组分容量及分配系数;(2)各组分的n及n eff;(3)各组分的H 值及H eff值;(4)画出四组分的K/值之间的关系曲线表3-1 在HPLC柱上测得的A、B、C、D 的组分t R(min) W h/2(min)⾮滞留组分ABCD 4.06.513.514.620.10.420.971.101.38答:(1)K/(A=0.60;B=2.38;C=2.65;D=4.03);(2)(A=4021;B=3099;C=2818;D=3394)n eff(A=595;B=1535;C=1486;D=2178);(3)H(A=0.06;B=0.08;C=0.09;D=0.07)H eff(A=0.42;B=0.16;C=0.17;D=0.11);(5)根据已有数据,绘出K/--n--n eff曲线,⾃⾏判断正确与否,并分析原因。


FID-2014检测器(毛细柱)基本操作步骤此基本操作步骤仅作参考使用,应以岛津说明书为准。
开机步骤1.根据要求配置好样品。
2.根据样品要求选择合适的柱子,并安装到SPL进样口及FID检测器上。
3.打开氮气源阀门,二次压力达到0.5 Mpa以上。
4.打开主机电源开关,在主机自检完后打开实时分析工作站。
5.点击“配置维护”(Configuration and Maintenance)图标,再点击“系统配置”(System Configuration)图标。
从中设置好分析流路(包括SPL进样口、色谱柱及FID检测器,如有其他配置还需选好)及相关参数。
6.点击菜单栏中的“视图”(View),选择“仪器监视器”(Instrument Monitor)。
在监视器窗口中确认a)载气(Carrier Gas)及吹扫流量(Purge Flow)为“打开”(ON)状态;b)FID检测器(Detector)及点火(Flame)为“关闭”(OFF)状态;7.点击“仪器参数”(Instrument Parameters)图标,a)将柱箱温度(Column)设置为30℃;b)将SPL进样口温度设置为40℃,并设置好SPL进样口中的柱流量(Column Flow)及分流比(Split Ratio)等参数;c)将FID检测器温度设置为40度,将FID检测器尾吹气(Make Up)压力表调整设置为75Kpa(30ml/min)d)点击“下载参数”(Download Parameters)图标。
8.点击“开启系统”(System ON)图标。
按仪器上的MONIT键。
9.设置FID温度、进样口温度为分析时的温度,点击“下载参数”(Download Parameters)图标,待FID检测器温度升至150℃以上时,在监视器窗口中打开FID检测器(Detector)开关为“打开”(ON)状态;10.打开氢气瓶阀门,二次压力达到0.3 Mpa;打开空气瓶阀门,二次压力达到0.3 Mpa;调整压力表,空气压力表为50KPa,氢气压力表为50KPa,再打开点火(Flame)开关,点燃火焰。
有机磷农药毛细柱气相色谱法(GC1.办法原理采纳二氯甲烷分三次萃取水样,用毛细柱GC-FPD分析测定有机磷农药含量。
2. 干扰及消退当水体中含有较多的有机物质时,萃取时激烈振荡会产生严峻的乳化现象,影响预处理操作,造成损失,添加适量的氯化钠可以避开产生严峻的乳化现象,以消退干扰。
3. 办法的适用范围本办法适用于有机磷农药厂排放的废水、地表水以及地下水中12种有机磷农药的测定,本办法的最低检出限为0.0l μg/L。
4.仪器①气相色谱仪,具火焰光度检测器。
②色谱柱:HR-1701石英毛细管色谱柱,25m×0.25mm(内径),0.25μm。
5. 试剂①二氯甲烷、丙酮,分析纯,色谱测定无干扰峰存在,否则需重蒸。
②无水硫酸钠、氯化钠,分析纯,450℃加热4h,无水硫酸钠冷却后保存在干燥器内。
③有机磷农药标准溶液,敌敌畏、二嗪农、异稻瘟净、乐果、甲基对硫磷、马拉硫磷、杀螟松、对硫磷、水胺硫磷、稻丰散、杀扑磷、乙硫磷标准溶液浓度均为100mg/L。
④试验用水为二次蒸馏水。
6. 步骤 (1)样品的采集与保存采集1000m1水样,储藏于棕色玻璃瓶,调整pH至6-7。
采集的水样若不准时测定,应置于4℃冰箱内保存。
大部分地表水中的有机磷会在14d内发生降解,因此在采样后7d内应对样品举行萃取,萃取液至多能保存14d,且应置于4℃冰箱内保存。
(2) 样品的预处理取500ml水样于1000m1分液漏斗中,调整pH6-7,加入25g氯化钠溶解后,加入60m1二氯甲烷,振荡萃取l0min,静置分层。
回收有机相,再用60m1二氯甲烷萃取一次,合并有机相。
有机相用无水硫酸钠干燥后,在旋转蒸发器内浓缩至4m1,再用高纯氮气吹至0.5m1。
(3)色谱条件色谱柱温度:170℃→15℃/min→210℃(1min)→10℃/min-220℃→15℃/min→240℃(5min)。
进样口温度:240℃;检测器温度:250℃。
第六章毛细管柱气相色谱法第一节毛细管气相色谱仪现代的实验室用的气相色谱仪大都既可用作填充柱气相色谱又可用作毛细管色谱仪。
毛细管色谱仪应用范围广,可用于分析复杂有机物,如石油成分,天然产物,环境污染,农药残留等。
图6-1是毛细管气相色谱仪示意图,与填充柱色谱仪比,毛细管色谱仪在柱前多一个分流-不分流进样器,柱后加一个尾吹气路。
由于毛细管柱体积很小,柱容量很小,出峰快,所以死体积一定要小,要求瞬间注入极小量样品,因此柱前要分流。
对进样技术要求高,对操作条件要求严。
尾吹的目的是减小死体积和柱末端效应。
毛细管柱对固定液的要求不苛刻,一般2-3根不同极性的柱子可解决大部分的分析问题。
毛细管柱一般配有响应快,灵敏度高的质量型检测器。
高分辨率毛细管气相色谱仪的三要素是:要选择好的毛细管柱及最佳分析条件;按样品选择合适的毛细管进样系统;选择高性能的毛细管气相色谱仪。
图6-1 毛细管气相色谱仪示意图第二节毛细管色谱柱1957年,美国科学家Golay提出毛细管柱的气相色谱法。
Golay称毛细管色谱柱为开管柱。
因这种色谱柱中心是空的。
毛细管柱是内径为Φ0.1-0.5mm左右、长度为10-300m的毛细柱,虽然每米理论板数约为2000-5000,与填充柱相当,但由于柱子很长,总柱效可高达106。
一、毛细管色谱柱组成通常来说,一根毛细管色谱柱由管身和固定相两部分组成。
管身采用熔融二氧化硅(熔融石英),通常在其表面涂上一层聚酰亚胺保护层。
涂层后的熔融石英毛细管呈褐色:但是涂层后的毛细管之间的颜色却不尽相同。
色谱柱的颜色对于其色谱性能没有什么影响。
经过持续的较高温度处理后.聚酰亚胺涂层管的的温度会变得比以前更深:标准的聚酰亚胺涂层管熔融石英管的温度上限为360℃,高温聚酰亚胺涂层管的温度上限为400℃。
固定相种类很多,大部分的固定相是热稳定性好的聚合物,常用的有聚硅氧烷和聚乙二醇。
另外还有一类是小的多孔粒子组成的聚合物或沸石(例如氧化铝、分子筛等)。
第六章毛细管柱气相色谱法第一节毛细管气相色谱仪现代的实验室用的气相色谱仪大都既可用作填充柱气相色谱又可用作毛细管色谱仪。
毛细管色谱仪应用范围广,可用于分析复杂有机物,如石油成分,天然产物,环境污染,农药残留等。
图6-1是毛细管气相色谱仪示意图,与填充柱色谱仪比,毛细管色谱仪在柱前多一个分流-不分流进样器,柱后加一个尾吹气路。
由于毛细管柱体积很小,柱容量很小,出峰快,所以死体积一定要小,要求瞬间注入极小量样品,因此柱前要分流。
对进样技术要求高,对操作条件要求严。
尾吹的目的是减小死体积和柱末端效应。
毛细管柱对固定液的要求不苛刻,一般2-3根不同极性的柱子可解决大部分的分析问题。
毛细管柱一般配有响应快,灵敏度高的质量型检测器。
高分辨率毛细管气相色谱仪的三要素是:要选择好的毛细管柱及最佳分析条件;按样品选择合适的毛细管进样系统;选择高性能的毛细管气相色谱仪。
图6-1 毛细管气相色谱仪示意图第二节毛细管色谱柱1957年,美国科学家Golay提出毛细管柱的气相色谱法。
Golay称毛细管色谱柱为开管柱。
因这种色谱柱中心是空的。
毛细管柱是内径为Φ0.1-0.5mm左右、长度为10-300m的毛细柱,虽然每米理论板数约为2000-5000,与填充柱相当,但由于柱子很长,总柱效可高达106。
一、毛细管色谱柱组成通常来说,一根毛细管色谱柱由管身和固定相两部分组成。
管身采用熔融二氧化硅(熔融石英),通常在其表面涂上一层聚酰亚胺保护层。
涂层后的熔融石英毛细管呈褐色:但是涂层后的毛细管之间的颜色却不尽相同。
色谱柱的颜色对于其色谱性能没有什么影响。
经过持续的较高温度处理后.聚酰亚胺涂层管的的温度会变得比以前更深:标准的聚酰亚胺涂层管熔融石英管的温度上限为360℃,高温聚酰亚胺涂层管的温度上限为400℃。
固定相种类很多,大部分的固定相是热稳定性好的聚合物,常用的有聚硅氧烷和聚乙二醇。
另外还有一类是小的多孔粒子组成的聚合物或沸石(例如氧化铝、分子筛等)。
熔融石英管的内表面会用一些化学方法进行处理,尽量的减小样品和管壁之间可能存在的相互作用。
所用的试剂和处理方法一般是依据将要涂在内壁上的固定相种类来确定的。
硅烷化处理则是最为常用的处理方式,使用硅烷类的试剂和管壁内表面上的硅基醇基团进行反应,使其变为甲基硅烷基或苯甲基甲基硅烷基。
当实验要求更高的使用温度时,我们可以来用不锈钢毛细柱来代替熔融石英毛细柱。
不锈钢毛细柱在使用温度(耐高温)及日常维护(不易折断等)的性能和指标上都优于熔融石英毛细柱。
但是不锈钢材质的惰性没有熔融石英好,它可以和许多的化合物相互作用,产生反应。
所以通常可以用化学方法对其进行处理,或者是在它的内壁再涂上薄薄的一层熔融石英,以增加不锈钢管的隋性:经过适当处理后,不锈钢毛细柱的惰性与熔融石英毛细柱的不相上下。
二、毛细管色谱柱固定相(一)气-液色谱固定相1.聚硅氧烷聚硅氧烷有优良的稳定性, 用途广,是目前最为常用的固定相。
标准的聚硅氧烷是由许多单个的硅氧烷重复联接构成,每个硅原子与两个功能基团相连,功能基团的类型和数量决定了固定相总体类型和性质,常见的四种功能基团为甲基、氰丙基、三氟丙基和苯基。
最基本的聚硅氧烷是由100%甲基取代的。
当有其他种类的取代基出现时,该基团的数量将由一个百分数来表示。
例如:5%二苯基—95%二甲基聚硅氧烷表示其包含有5%的苯基基团和95%的甲基基团。
“二”是表示每个硅原子包含有两个特定基团,但当两个特定基团完全相同时,我们有时也会省略这种叫法。
如果甲基的百分数没有表征,则表示它的含量可能是100%(如50%苯基—甲基聚硅氧烷表示甲基的含量为50%)。
有时我们可能对氰丙基苯基的百分含量产生错误的理解,如14%氰丙基苯基—二甲基聚硅氧烷表示的是其含有7%氰丙基和7%苯基(另有86%的甲基),因为一个氰丙基和一个苯基连接于同一个硅原子上,所以14%是一种加和的表征方式。
我们有时会用低流失来表征一类固定相。
这一类固定相是在硅氧烷聚合物中链接一定数量的苯基或苯基类的基团,通常我们称之为“亚芳基”。
由于它们的加入,聚合物的链接变得更加坚固稳定,保证了在较高温度时,固定相不会产生降解。
也就是说,进一步降低了色谱柱的柱流失,提高了色谱柱的使用温度。
与原始的非亚芳基类型的固定相相比,亚芳基固定相不仅拥有相同的分离指数,而且在色谱柱的维护等方面也有许多的调整(例如SE-52和SE-54)。
尽管同类普通型和低流失型固定相的分离性能相同或极为相似,但是在某些方面还有微小的区别。
另外,我们也使用一些独特低流失固定相。
2.聚乙二醇聚乙二醇是另外一类广泛应用的固定相。
有时我们称之为“WAX”或“FFAP”。
聚乙二醇不像聚硅氧烷那样有多种取代基团,它是100%固定基质的聚合物。
相对于聚硅氧烷,聚乙二醇固定相色谱柱的寿命较短,而且容易受温度和环境(有氧环境等)的影响。
另外,聚乙二醇固定相在相应的GC实验条件下需保持液态。
但由于其独特的分离性能,聚乙二醇仍是我们常用的固定相之一。
常用的聚乙二醇GC固定相有两种:一种是能在较高温度下使用的,但是它的活性相对较高一些(也就是说有些化合物的色谱峰会有拖尾现象);另一种的使用温度上限较低,温度下限也较低,但使用中所表现出的再现性和惰性比上一种要好。
在分离指数上,上述两种固定相有轻微的差异。
还有一种是pH阳离子改性聚乙二醇固定相。
FFAP柱就是一类用对苯二甲酸改性的聚乙二醇作为固定相的。
这种色谱柱常用于分离分析酸性化合物。
另外,我们也用碱性化合物对聚乙二醇固定相改性用来分离分析碱性化合物(CAM)。
普通分析色谱柱分离强酸或强碱化合物时会出现色谱峰拖尾现象,使用pH 改性固定相后,这种现象会明显地减小。
(二)气-固色谱固定相就是在管壁表面粘合很薄一层的小颗粒物质,通常叫做多孔层开口管(PLOT)柱。
样品是通过在固定相上产生吸附/脱附作用来分离的。
最为常用的PLOT柱固定相有苯乙烯衍生物、氧化铝和分子筛等。
PLOT柱的保留性能非常突出,用它可以进行那些常规固定相做不到的分析分离。
对于那些要求在低于室温的条件下,使用聚硅氧烷或聚乙二醇固定相进行的分析分离,PLOT柱在室温或高于室温的状态下就可以轻易完成。
烃类和硫化物气体、惰性和永久性气体以及低沸点溶剂等都是常用PLOT 柱进行分析分离的化合物。
(三)键合和交联固定相交联是将多个聚合物链单体通过共价键进行连接,键合是将其再通过共价键与管壁表面相连。
这样处理的结果使得固定相的热稳定性和溶剂稳定性都有较大的提高。
所以,键合交联固定相色谱柱可以通过溶剂的浸洗,从而去除柱内的污染物。
大多数的聚硅氧烷和聚乙二醇固定相都是经过键合交联处理的。
另有少数固定相是不用键合或键合交联进行处理的。
但如有可能,能够进行键合交联的,都会对固定相做出相应的处理。
三、柱流失所有的色谱柱都有柱流失的现象。
这是由于固定相的正常降解而产生的被洗脱物质。
柱流失会随着温度的升高加剧。
我们可以通过流失曲线或图清楚地看到这种变化。
一般我们会在程序升温的条件下做一次空白试验,温度要升至色谱柱的温度上限,并持续该温度10-15分钟,这样就可以得到该色谱柱的正常流失曲线图。
从流失图中我们可以得到几个重要的指标。
空白试验的基线在较低温度区域相对平坦,到离温度上限30-40℃时开始急速地上升,直至达到温度上限。
在上限温度持续期间,基线又变得平稳许多。
几分钟后基线会又变得完全平坦。
如出现明显或严重的偏差,并不是由于色谱柱流失引起的。
色谱柱的流失是一种持续的过程,并不会偶然地开始,也不会突然地停止。
如果在空白试验中得到了色谱峰,这并不是由于柱流失而引起的,它极有可能是GC系统中的污染物质。
使用质谱检测器进行检测并与谱图库对照,您会发现它们是一些含硅的化合物。
它们的来源极有可能是进样垫。
一般来说,极性固定相的流失率较高,较低温度下,它们的流失就很明显。
如果您使用的检测器对固定相中任何原子或功能团都有特别灵敏的响应,那么柱流失就非常明显了。
就算柱流失不是很严重,但由于检测器对柱内降解产物有较灵敏的响应,会导致很强的基线噪声。
在氰丙基取代聚硅氧烷固定相与NPD检测器系统或聚乙二醇柱与ECD检测器系统中,这种现象就很突出。
由流失图中我们可以看到,在高温区域柱流失会迅速升高。
当流失率增高时,我们无法用一种绝对的方法去测量指示。
柱流失最佳的测量方法是测量在两种温度下背景信号的不同或改变。
通常我们会选择色谱柱的温度上限和100℃这两个点,绝对的背景信号通常是整个GC系统背景的组合,我们不可能测量出柱流失对这个信号有多大的贡献。
而测量柱流失的相对数量,其它对背景信号有贡献的因素也就被减去了。
大多数的色谱柱是通过FID进行检测的。
FID的输出信号为微微安培(pA)。
流失水平就是在两种温度下FID信号值的差(ΔpA)。
由于这些数值随检测器响应的变化而变化,所以只有在相同的实验条件下使用同一个检测器,或者在标准的流量条件下使用相同标准的检测器,并且流失数值以pg /克固定相来表示,这样做的数据才真实有效。
随着色谱柱的使用,柱流失会不断地升高。
色谱柱暴露于有氧环境(空气)中和持续在等于或接近色谱柱的上限温度条件下被使用,都会加速色谱柱的流失。
柱流失突然或快速的升高则可能是色谱柱。