材料设计—14-一维单原子链振动
- 格式:ppt
- 大小:992.00 KB
- 文档页数:32
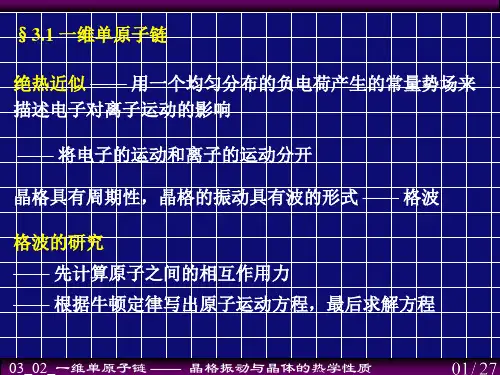

一维单原子链晶格振动解析步骤一维单原子链模型是固体物理中的经典模型之一,用于描述晶体中原子的振动行为。
在这个模型中,原子由质量为m的核和劲度系数为K的弹性相互作用构成。
通过对一维单原子链的晶格振动进行分析,可以更好地理解固体中的声子模式和声子色散关系。
下面将介绍一维单原子链晶格振动解析步骤:第一步:建立模型首先,我们要建立一维单原子链的模型。
假设晶格常数为a,原子间距为a/2,一维晶格中的每个原子都沿着x轴定位。
原子间的相互作用由弹簧模型描述,即相邻原子间的相互作用劲度系数为K。
这个模型是一个简单的原子链模型,可以通过它来研究晶格振动的基本性质。
第二步:求解运动方程接下来,我们需要求解原子在这个一维单原子链中的运动方程。
假设第n个原子的位移为Un(t),那么根据牛顿第二定律,可以得出该原子的运动方程为:m*Un’’(t) = -K*(Un(t+0) - 2*Un(t) + Un(t-0))上式中,Un’’(t)表示Un对时间的二阶导数,-K*(Un(t+0) -2*Un(t) + Un(t-0))表示受到的弹性相互作用力。
第三步:假设解的形式由于原子在一维单原子链中的振动属于谐振动问题,我们可以假设原子的位移满足解的形式为:Un(t) = An*exp(i*(k*n*a - ω*t))其中,An是振幅,k是波数,ω是角频率,n是原子的编号。
将这个解代入到运动方程中,可以得到关于角频率ω和波数k的关系式,即声子色散关系。
声子色散关系描述了声子的能量随波数变化的关系,是描述晶体中声子性质的重要工具。
第四步:得到声子色散关系将解的形式代入运动方程,我们可以得到关于角频率ω和波数k的关系式。
具体地,我们可以得到一维单原子链中的声子色散关系为:ω(k) = 2*sqrt(K/m)*|sin(ka/2)|声子色散关系描述了一维单原子链中的声子能量随波数变化的规律。
从这个关系式可以看出,一维单原子链中的声子有声学支和光学支两种振动模式,它们的能量随波数的变化方式不同。

一维原子链的晶格振动方程晶体是由原子或分子组成的周期性排列的结构,其内部的原子或分子通过振动相互作用,从而产生晶格振动。
晶格振动方程描述了一维原子链中原子的振动行为,对研究固体物理和材料科学具有重要意义。
一维原子链的晶格振动方程可以通过简化模型来描述。
我们假设原子链中的原子质量相同,且原子间的相互作用力为弹簧力。
在平衡位置附近,原子的位移可以用小量近似表示,即位移量远小于原子间距。
此时,可以利用胡克定律,将原子间的相互作用力近似为线性弹簧力。
根据胡克定律,弹簧的力与其伸长(或缩短)的长度成正比,且方向与伸长(或缩短)的方向相反。
对于一维原子链中相邻两个原子,其相互作用力可以表示为:F = -k(x - a) - k(x + a)其中,F为相互作用力,k为弹簧常数,x为原子的位移量,a为原子间距。
根据牛顿第二定律,物体的加速度与作用力成正比,与物体的质量成反比。
在一维原子链中,每个原子的加速度与相邻原子的相互作用力有关,可以表示为:m(d^2x/dt^2) = -k(x - a) - k(x + a)其中,m为原子的质量,d^2x/dt^2为原子的加速度,t为时间。
将上述方程进行简化,可得:d^2x/dt^2 = -k/m * (2x - a - a)化简后得到:d^2x/dt^2 = -2k/m * (x - a/2)这就是一维原子链的晶格振动方程。
从中可以看出,原子的加速度与位移量成正比,且与原子间距和弹簧常数有关。
当原子受到外力作用时,晶格振动方程可以进一步进行修正。
一维原子链的晶格振动方程可以通过求解微分方程得到解析解,也可以通过数值模拟方法进行计算。
对于周期性边界条件下的一维原子链,可以采用傅里叶变换的方法,将晶格振动分解为一系列特定频率的波动模式。
晶格振动在固体物理和材料科学中具有广泛的应用。
通过研究晶格振动,可以揭示物质的热力学性质、电子结构和传热传电等基本行为。
此外,晶格振动还与声学性质、热导率、热膨胀和热容等宏观性质密切相关,对于材料的设计和优化具有重要意义。


一维单原子链的频率分布一维单原子链是指由相同类型的原子按照一定的规则排列成的链状结构。
频率分布是指在单原子链中各个振动模式的频率出现的分布情况。
本文将从单原子链的基本特征、频率的计算方法以及频率分布的特点三个方面来详细探讨一维单原子链的频率分布。
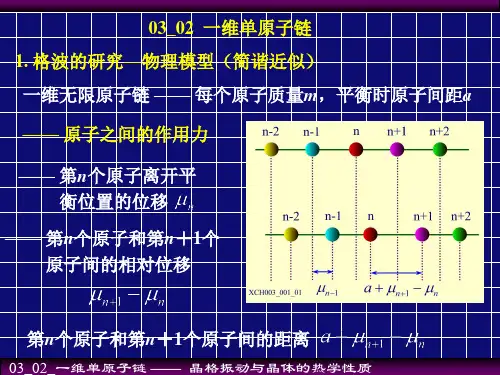
一、单原子链的基本特征一维单原子链是凝聚态物理中常见的模型系统,它具有以下基本特征:1. 原子之间的相互作用力:在单原子链中,相邻原子之间存在着弹性力和相互作用力,这些力决定了原子在链中的振动行为。
2. 间距和质量的均匀性:单原子链中的原子间距相等,原子质量也相等,这使得单原子链具有均质性,便于分析和计算。
3. 边界条件:单原子链的两端通常会施加边界条件,如固定边界条件或周期性边界条件,以模拟实际情况中的约束条件。
二、频率的计算方法在一维单原子链中,原子的振动可以通过离散化简化为谐振子模型,通过求解谐振子的本征值问题可以得到振动频率。
对于一维单原子链,振动频率的计算方法如下:1. 利用牛顿第二定律:应用牛顿第二定律,可以得到原子的运动方程。
通过求解运动方程可以得到振动频率。
2. 应用弹性势能:利用弹性势能的定义,可以将原子的振动视为在势能函数中寻找最小值的过程。
通过求解势能函数的最小值问题,可以得到振动频率。
3. 应用量子力学:在一维单原子链中,可以将原子的振动量子化,利用量子力学的方法求解振动频率。
具体的计算方法可以通过哈密顿算符的对角化来实现。
三、频率分布的特点在一维单原子链中,频率分布具有以下特点:1. 频率的离散性:由于单原子链的离散结构,振动频率呈现出离散的特点。
频率分布通常由一系列离散的振动模式组成,每个模式对应一个特定的频率。
2. 频率的对称性:对于一维周期性边界条件的单原子链,频率分布具有对称性。
即频率分布在频率为零的点处对称,且对称轴上的频率相等。
3. 频率的分布范围:频率分布的范围取决于原子之间的相互作用力和边界条件。
不同的相互作用力和边界条件将导致不同的频率分布范围。



第n-2个原子第n-1个原子第n+1个原子第n+2个原子第n个原子maµn-2µn-1µnµn+1µn+2第n-2个原子第n-1个原子第n+1个原子第n+2个原子第n个原子maµn-2µn-1µnµn+1µn+2a一维晶格仅考虑最近邻原子间相互作用时的色散关系qv p ω=2.21∑=qqQ 221∑⎟⎠⎞⎜⎝⎛=•n n m T μ∑••=qiqna q n t Q Nmt ,)e (1)(μ∑∑∑′−′−′=nq qinaq q .q ina q .,t Q t QN T )e ()e (21∑∑∑′+′−′=q nq q ina qq .q .,Nt Q t Q)(e1)()(21∑∑′−′′=q qqq qq t Qt Q,)()(21,..δ∑−=qqqt Qt Q)()(21..∑=qqqt Qt Q)()(21.*.)()(*t Q t Q q q =−动能的正则坐标表示:势能∑−=qinaqq n eQ Nm 1μ∑−−−='')1('11q aq n i q n e QNm μ∑−−=nn n U 21)(21μμβ1(')'(')','1{[1]}()2N ia q q iaq iaq ina q q q q q q n U Q Q e e e emNβ−++==+−−∑∑}2{2∑−−−−=qiaq iaq qq e e Q Q mβ{1cos()}q qqQ Qaq mβ−=−∑代入上式,得:*{1cos()}qqqU Q Q aq mβ=−∑利用)}cos(1{22aq mq −=βω2*12q q q qU Q Q ω=∑2221∑=qq q Q U ω系统势能所以2221∑=qq q Q U ω哈密顿量2221()2q q qqH T U Q Q ω=+=+∑ ——系统复数形式的简正坐标ti q q q eA Nm Q ω=势能动能∑=qq Q T 221 1()[()()]2Q q a q ib q =+)]()([21)(*q ib q a q Q −=∑=qq Q T 2212221∑=qq q Q U ω∑>+=22)]()([21q q b q a T 实数形式的简正坐标令∑>+=222)]()([21q q q b q a U ω能量本征值qq n n qωε=)21(+=2()/exp()()2qq n q q n Q H ξϕωξ=−=本征态函数一个简正坐标对应一个谐振子方程,波函数是以简正坐标为宗量的谐振子波函数。

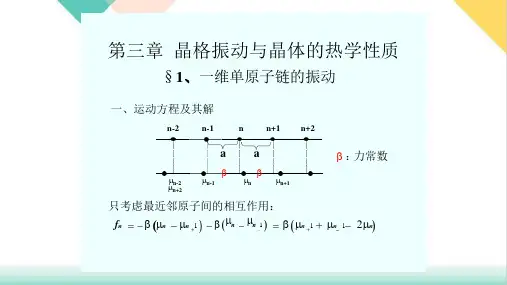
第九讲:晶体振动上一维单原子链简谐近似和简正坐标布拉伐晶格晶体中的格点表示原子的平衡位置,原子在格点附近作热振动,由于晶体内原子之间存在相互作用力,各个原子的振动不是孤立的,而是相互联系在一起的,因此在晶体中形成各种模式的波,称为格波。
只有当振动非常微弱时,原子间的相互作用可以认为是简谐的,非简谐的相互作用可以忽略,在简谐近似下,振动模式才是独立的。
由于晶体的平移对称性,振动模式所取的能量值不是连续的,而是分立的。
通常用一系列独立的简谐振子来描述这些独立的振动模,它们的能量量子称为声子。
势能和动能函数设简单晶格晶体包含N 个原子,平衡位置为R n ,偏离平衡位置的位移矢量为µn (t ),则原子的位置为()()R R n n n t t '=+µ。
将位移矢量µn (t )用分量表示,写成µi ( i = 1, 2, ..., 3N )。
N 个原子体系的势能函数可以在平衡位置附近展开成泰勒级数:⋅⋅⋅++ +=∑∑==j i N i N j i j i i i V V V V µµ∂µ∂µ∂µ∂µ∂03131,20021 (3-1) 下标0表示为在平衡位置时所具有的值。
可以设V 0 = 0,而且在平衡位置相互作用力为零:0 0=i V ∂µ∂ (3-2) 忽略二阶以上的非简谐项可得:j i N j i ji V V µµ∂µ∂µ∂031,221∑==(3-3) N 个原子体系的动能函数为:∑==Ni ii m T 31221µ(3-4)简正坐标 为了使问题简化,引入简正坐标N Q Q Q 321 , , ,⋅⋅⋅简正坐标和原子的位移坐标 µi 之间通过正交变换相互联系:∑==Nj jij i i Qa m 31µ (3-5)引入简正坐标后体系的势能函数和动能函数为:∑==Ni iQT 31221(3-6)∑==N i ii QV 312221ω (3-7)由于动能函数T 是正定的,根据线性代数的理论,总可以找到这样的正交变换,使势能函数和动能函数同时化为平方项之和。
一维单原子链振动能与热容的研究刘凤智【摘要】对于一维晶格可以严格求出振动能和热容,将它们与爱因斯坦模型和德拜模型的结果进行比较,可以找出两种模型的成功与不足,如此可以加深对模型的理解,也有助于模型的完善.【期刊名称】河南科学【年(卷),期】2012(030)009【总页数】5【关键词】单原子链;振动能;热容;爱因斯坦模型;德拜模型晶格振动理论是晶体的重要理论,从晶格振动理论可以推出晶体的物理性质,特别是晶格热容的计算.对于一维晶格振动,因为模型简单,常常可以得出严格解,而受到人们的重视,一维单原子链也是晶格振动理论的入门知识,在固体物理教学中占有重要的地位.晶格振动理论就是起源于对晶格热学性质的研究,而能量和热容是热学性质中很有代表性的物理量.对晶格热容和振动能的具体求解是一个比较复杂的问题,在一般的讨论中常采用爱因斯坦和德拜两个简化模型.因为一维单原子可以做严格意义上的理论研究,为了深刻理解模型的实质,可以对一维晶格进行对比研究,找出爱因斯坦和德拜模型的成功与不足,有利于对实际三维晶格的认识[1].本文首先从理论上将三种处理方法应用到一维单原子链,得出相应的振动能与热容,并得出极端情况下的极限值,随后用一个实际例子做了讨论,得出一些有意义的结论.1 由爱因斯坦模型计算晶格振动能和等容摩尔热容对于一维单原子链,假设原子数为N,晶格常数为a,晶格的力常数为β,原子质量为m,只考虑最近邻原子的作用,可以得出晶格振动的色散关系[2]:其中:ωm为晶格振动的截止频率;q为波矢,可以将它限制在第一布里渊区,即-π/a<q≤π/a.进一步假设晶体为绝缘体(不考虑电子运动的贡献),则一维晶格振动能为:如果采用爱因斯坦模型,即假设所有原子都以统一的频率ωE(称为爱因斯坦特征频率)振动,则一摩尔晶格振动能为:其中为了方便可以引入一个特征温度,称为爱因斯坦温度:爱因斯坦温度θE,一般由实验确定.上述晶格振动能的极限情况:由摩尔热容的定义可以从(3)式求出晶格的等容摩尔热容:等容摩尔热容的极限情况:2 由德拜模型计算晶格振动能和等容摩尔热容将德拜方法用到一维晶格振动,首先考虑长波近似下的色散关系[3]:此时可以求出频谱密度为:还可以求出德拜截止频率:由德拜模型可以求出一维晶格的摩尔振动能为:其中称为德拜温度,一般需要由实验来确定:对于德拜模型的一维晶格振动能的极限情况:由德拜模型求出晶格的等容摩尔热容:对于德拜模型一维晶格等容摩尔热容的极限情况:3 由严格解法求晶格振动能和等容热容对于一维晶格可以求出严格的频谱密度:由(16)式和振动能公式可以得出一维晶格摩尔振动能:其中为了方便还可以定义一个特征温度由严格频谱密度求出晶格摩尔振动能的极限情况[4]:由(17)式还可以求出相应的等容摩尔热容:等容摩尔热容的极限情况:4 讨论4.1 三种振动能的比较假设一维单原子链中振动截止频率为ωm=4.479×1013(rad/s),可分别计算出德拜模型的特征温度θD=535.3 K,严格计算中的特征温度θm=340.8 K.下面根据室温下,由爱因斯坦模型中的振动能(3)式和严格解法的振动能(17)式,二者能量应该相等,即有:将θm=340.8 K和T=300 K,代入(21)式中,通过数值计算,可得下面的方程:从(22)式可解得爱因斯坦温度:θE=232.4 K.知道了特征温度,通过数值计算可以分别求出各自的振动能,其振动能随温度的变化曲线如图1所示.由三条能量曲线看,在温度较高时,能量与温度基本成线性增长,随着温度的增长,三条能量曲线趋于重合,与经典能均分定理的结论一致,量子效应消失,这也说明在高温区,三种模型都能很好地描述晶格振动能.由图1所示,在低温区,三条曲线差异较大.由爱因斯坦模型得出的能量在75 K以下的低温区偏离严格解法所得能量,在20 K左右就基本接近零点能了,不过二者的零点能相差只有7%左右.对于德拜模型与严格解法二者的曲线在低温区差异较大,特别是在零点能,二者差距达到23%之多,这是由于德拜近似将截止频率抬高了的缘故,由于德拜近似主要用来计算晶格比热,对于振动能不太关注,但有时在计算结合能时,会用它来计算零点振动能,此时会有较大误差,应该引起重视.顺便提及,由数值计算得出的零点能与各个低温极限所给出的结果完全相符,这也说明各个极限公式是正确的.4 .2 三种等容摩尔热容的比较确定了各自的特征温度,也可以通过数值计算来研究等容摩尔热容与温度的关系.计算所得等容摩尔热容随温度变化的曲线如图2所示.我们依然将严格解法所得的结果作为准确值,用它来比较爱因斯坦和德拜模型.如图2所示,在高温区,德拜模型与爱因斯坦模型都与严格解相同,说明在高温情况下,二种模型都能给出正确的晶格热容,也与经典物理理论相符.在低温区,爱因斯坦模型与严格解相差较大,在100 K左右开始偏离,在20 K左右就基本下降为零了,下降速度远快于温度的一次方.这与爱因斯坦模型过于简单有关,爱因斯坦模型认为所有振动都以一个单一频率ωE进行,没有考虑频率的分布.在温度很低时德拜模型与严格解法都得出等容摩尔热容与温度呈线性下降,这段温区很小,从表1可以看出,较严格的线性关系在10 K左右(误差在0.5%以内),这与文献[5]的说法相符(从一维的结论来推知三维的实际晶体,在低温时热容与温度的3次方下降的规律可能只在很小的一个温区成立),大约为θD/50的量级.从比热随温度变化的数据曲线上来看,虽然高温极限与低温极限德拜模型都与严格解相符,但在中低温区,二者有较大的差异,这也可能是在实际晶体中德拜模型计算热容相差较大的原因.在高温区爱因斯坦等容摩尔热容曲线是在德拜等容摩尔热容曲线的上方,这一点与文献[5]的说法不同.5 结论我们从一维单原子为研究对象,将爱因斯坦模型和德拜模型与严格解进行了比较,清晰地指出了两种常见模型的成功与缺陷,主要得出以下几个结论:1)可以用能量相等的方法来确定爱因斯坦温度.2)用德拜模型来求零点能会产生较大的误差.3)对于一维单原子链,晶格热容随温度线性下降的温区比预料的要小得多. 4)在高温区,爱因斯坦热容曲线在德拜曲线的上方,在低温区则在下方.5)尽管德拜热容曲线在极低温与高温区与严格解相符,但在中温区曲线下移,这有可能是导致与实际热容偏差的原因.参考文献:[1]张海峰,徐文兰.几种一维原子链晶格振动特性比较研究[J].中国科学院研究生院学报,1995,12(1):86-90.[2]方俊鑫,陆栋.固体物理学:上册[M].上海:上海科学技术出版社,1981.[3]胡安,章维益.固体物理学[M].北京:高等教育出版社,2005.[4]刘丹.基于德拜模型讨论晶格比热[J].湖北教育学院学报,2007,24(8):10-12.[5](美)德克尔A J.固体物理学[M].高联佩,译.北京:科学出版社,1965.(编辑张继学)基金项目:辽宁石油化工大学科研基金项目(80040067)。