单晶结构解析技巧
- 格式:doc
- 大小:243.50 KB
- 文档页数:11


有关单晶培养的问题 1.单晶培养的方法多种多样,我们没必要掌握那些难以操作的,如升华法、共结晶法等。
最简单的最实用。
常用的有1.溶剂缓慢挥发法;2.液相扩散法;3.气相扩散法。
99%的单晶是用以上三种方法培养出来的。
2.单晶培养所需样品用量 一般以10-25mg为佳,如果你只有2mg左右样品,也没关系,但这时就要选择液相扩散法和气相扩散法,不能使用溶剂缓慢挥发法。
3.单晶培养的样品的预处理 样品溶解后一定要过滤,不能用滤纸,而是用一小团棉花轻轻的塞在滴管的中下部或下部,不要塞太紧,否则流的太慢。
样品当然是越纯越好,不过如果实在没办法弄纯也没关系,培养一次就相当于提纯了一次,我经常用一些TLC显示有杂点的东西长单晶,但得多养几次。
4.一定要做好记录 一次就得到单晶的可能性比较小。
因此最好的方法就是在第一次培养单晶的时候,采取少量多溶剂体系的办法。
如果你有50mg样品,建议你以5mg为一单位,这样你可以同时实验10种溶剂体系,而不是选两种溶剂体系,每个体系25mg。
这是做好记录就特别重要,以免下次又采用已经失败的溶剂体系,而且单晶解析时必须知道所用的溶剂。
5.培养单晶时,最好放到没人碰的地方,这点大家都知道。
我想说的是你不能一天去看几次也不能放在那里5,6天不管。
也许有的溶剂体系一天就析出了晶体,结果5天后,溶剂全干了。
一般一天看一次合适,看的时候不要动它。
明显不行的体系(如析出絮状固体)就要重新用别的溶剂体系再重新培养。
6.液相扩散法中良溶剂与不良溶剂的比例最好为1:2-1:4。
7.烷基链超过4个碳的很难培养单晶。
8.分子中最好不要有叔丁基,因为容易无序,影响单晶解析的质量。
9.含氯的取代基一般容易长单晶,如4-氯苯基取代化合物比苯基取代化合物容易长单晶。
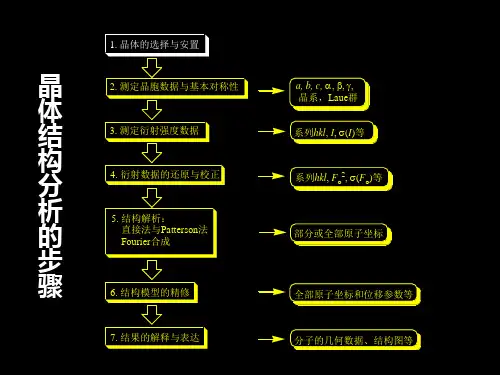
晶体结构解析步骤。
一支管法:在单晶制备时,经常会发现配位一发生,产生大量的微晶,再去挥发母液,怎么都长不大,以前听人说可用扩散法,但受到文献启示,可以找一根长玻璃管,底下注入盐的溶液,上面加一个纯溶剂缓冲层(可长可短),最上面注入(先慢后快)配体溶液,两三个小时或两三天就搞定了。

单晶结构解析单晶结构解析是指通过实验和计算,确定一种物质的单晶体结构及其晶体学参数的过程。
单晶结构解析对于物质的性质,结构及其在材料科学中的应用具有重要的意义。
下面将从实验过程、数据处理及结果分析三方面对单晶结构解析进行详细描述。
实验过程在进行单晶结构解析之前,需要先获得单晶样品。
获得单晶样品的方法主要包括晶体生长、晶体分离等。
单晶样品的获得需要具备一定的技术储备和经验。
一般情况下,单晶样品的获得需要先从大的多晶体中选择适合的晶体,再通过化学处理、物理处理等方法制备单晶样品。
获得单晶样品后,需要对其进行结构分析。
实验过程主要包括X射线单晶衍射实验、数据采集等步骤。
X射线单晶衍射实验是获得单晶结构信息的主要实验方法。
实验过程中需要将单晶样品置于X射线衍射仪中,然后进行数据采集。
根据实验条件和单晶样品的性质,可以选择不同类型的衍射仪,如旋转衍射法、Laue法等。
数据采集后,需要对数据进行处理。
数据处理数据处理是单晶结构解析的重要环节之一。
在数据处理过程中,需要消除噪声,确定有效数据。
常用的数据处理方法包括数据维护(检查数据质量)、数据分类、数据索引等方法。
数据维护是指检查数据质量,删除无效数据和不符合要求的数据。
数据分类是将有效数据根据其类型和强度进行分类和编码,为后续数据索引做准备。
数据索引是通过将不同类型的有效数据进行比对,旋转和移动,找出相应的基本数据并确定晶系、晶胞等结构参数。
结果分析单晶结构解析的最终结果是通过计算获得的晶胞参数,通过这些参数可以确定晶体的空间对称性、原子类型和位置等结构信息。
对于单晶结构解析结果的评价,需要考虑各种因素,如数据质量、数据采集方法、计算方法等。
评价单晶结构解析合理性的指标主要包括R值、Rfree值等。
R值是实验数据与模型预测之间差异的程度,R值越小说明模型和衍射数据之间的匹配越好。
Rfree值是根据实验数据和模型计算的一组独立数据与模型预测之间的差异,用于评估模型的过拟合程度。

单晶结构解析讲义完美版晶体结构解析1.Shelxtl 使⽤流程※解析原始⽂件有hkl⽂件(或raw⽂件),包含衍射数据;p4p⽂件,包含晶胞参数※为⼀个晶体的数据建⽴project,该项⽬下所有⽂件具有相同的⽂件名;⼀旦在XPREP 中发⽣hkl⽂件的矩阵转换,则需要输出新⽂件名的hkl等⽂件,因此要建⽴新的project。
※⾸先运⾏XPREP,寻找晶体的空间群※然后运⾏XS,根据XPREP设定的空间群,寻找结构初解※在Xshell中观察初解是否合理,如不合理,需重回XPREP中设定其他的空间群2.Xshell 使⽤流程※找出重原⼦或者确定性⼤的原⼦※找出其余⾮氢原⼦※精修原⼦坐标※精修各项异性参数※找到氢原⼦(理论加氢或差值傅⾥叶图加氢)※反复精修,直到wR2等指标收敛。
最后的R1<0.06(0.08) wR2<0.16(0.18)※通过HTAB指令寻找氢键,判定氢的位置是否合理,并且将相关氢键信息通过HTAB和EQIV指令写进ins⽂件中※将原⼦排序(sort)3.cif ⽂件⽣成和检测错误流程※在步骤1、2完成后,在ins⽂件中加⼊以下三条命令bond $Hconfacta※此时⽣成了cif和fcf⽂件,将cif⽂件拷贝到planton所在⽂件夹中检测错误,也可以通过如下在线检测⽹址:/doc/aaaed6d749649b6648d74737.html /services/cif/checkcif.html※根据错误提⽰信息,修改或重新精修,将A、B类错误务必全部消灭,C类错误尽量消灭。
4.Acta E 投稿准备流程投稿前,请务必切实做好如下⼯作:※按步骤1、2、3解析晶体并⽣成相应cif和fcf⽂件。
※准备结构式图(Chemical structural diagram)、分⼦椭球图(Molecular ellipsoid diagram)和晶胞堆积图(Packing diagram),最好是pdf格式。


单晶结构解析技巧1. 通常,H原子的处理方法作者要给出:(1)一般通过理论加H,其温度因子为固定值,可通过INS等文件查看(2) 水分子上H原子可通过Fourier syntheses得到(3)检查理论加上的H原子是否正确,主要看H原子的方向。
若不正确则删去再通过Fourier syntheses合成得到(4) 检查H原子的键长、键角、温度因子等参数是否正常。
通过检查分子间或分子内的H键是否合理最易看出H键的合理性(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)(6) 各位来说说方法与心得?2.胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio222_ALERT_3_B Large Non-Solvent H Ueq(max)/Ueq(min) ... 4.97 Ratio342_ALERT_3_B Low Bond Precision on C-C bonds (x 1000) Ang (49)B 级提示当然得重视了。
建议你先把H撤消,精修到C的热椭球不太变形和键长趋正常。
如做不到就要看空间群?衍射点变量比太小?以至追查到原始数据的录取参数和处理等。
这些粗略意见仅供参考,如何?3.在XP中画图时,只有一部分,想长出另外的对称部分。
我是envi完了,然后sgen长出来的,可是和symm显示的对称信息不一样。
比如:我根据envi的结果用sgen O1 4555得到的是O1A而不是O1D,这跟文献中标注的不一样啊,怎么统一呢?很困扰,忘达人指教。
xp里是按顺序编号的,第一个sgen出的的统一为A,依次标号。
你如果想一开始就统一D的话,重新name一下4.高氯酸根怎么精修呀?我用的SHETXL6.1版的,最好告诉我怎么用其中的XSHELL来做,我觉得他好用!Method 1DFIXDfix 1.42 0.02 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Dfix 1.42 0.02 O1 O2 O1 O3 O1 O4 O2 O3O2 O4O3 O4Method 2SADISadi 0.01 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Sadi 0.01 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O45. 晶体的无序是怎么造成的呀,是晶体培养的问题吗?如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5大于0.5没什么的,解完后都在1以下就可以了。


单晶培养及结构解析一、单晶培养1. 啥是单晶培养呢?哎呀,这就像是在微观世界里精心培育小宝贝一样。
你知道晶体吧,那些规则的、亮晶晶的东西。
单晶呢,就是由一颗晶核慢慢长大形成的单晶体。
单晶培养的方法有好多哦。
比如说溶液法,就像我们冲糖水一样,把溶质溶解在溶剂里,然后通过控制温度、浓度这些条件,让晶体慢慢长出来。
还有熔体法,这个就像是把固体加热变成液体,再让它慢慢冷却结晶。
不过呢,这些方法可都不简单,每一步都要特别小心。
2. 在溶液法中,选择合适的溶剂超级重要。
这就好比给小种子找合适的土壤。
如果溶剂不合适,晶体可能就长不好或者根本长不出来。
比如说有些物质在水里溶解性很好,但是在酒精里就不太行,所以要根据物质的性质来挑选溶剂。
而且,溶液的浓度也要恰到好处。
太稀了,晶体长出来的速度慢得像蜗牛爬;太浓了,又可能一下子就析出很多小晶体,而不是我们想要的大单晶。
3. 温度也是个关键因素。
就像人生活的环境温度一样,晶体生长也有它喜欢的温度范围。
如果温度波动太大,晶体可能就会长得歪歪扭扭的,就像我们被风吹得东倒西歪一样。
所以在培养单晶的时候,要把温度控制得稳稳的,有时候甚至需要用到恒温设备呢。
二、结构解析1. 晶体长出来了,我们还得知道它的内部结构呀。
这就像是要了解一个人的内心世界一样。
结构解析就是要搞清楚晶体里原子、分子是怎么排列的。
我们可以用X - 射线衍射技术来做这件事。
这个技术可神奇了,它就像一双超级透视眼,能够看到晶体内部原子的排列情况。
2. 当X - 射线照射到晶体上的时候,会发生衍射现象。
这些衍射图案就像是晶体内部结构的密码一样。
我们通过分析这些衍射图案,就能得到晶体的结构信息。
不过呢,这个分析过程可不容易,就像解密一样,需要用到很多复杂的数学公式和专业的软件。
3. 解析出晶体结构有什么用呢?这用处可大啦。
比如说在药物研发方面,如果我们知道了药物晶体的结构,就能更好地理解药物的性质,从而改进药物的效果。

shelxtlopen newnamexpfmolkill $qprojselect the good directionexittelp 0 -30plotfile enter file namedraw file nameselect file(ps file)black and whitecellfmolkill $qmatr 1=a 2=b 3=cpbox 5 15packselect (space=keep, enter=del) fmoltelp cellenter file namedraw file nameselect file type(a=psfile) black and white(enter)planexpread file namefmolmpln atom1 atom 2.....enteranglexpread file namefmolmpla n(atom number) atom1 atom 2.....mpla n(atom number) atom1 atom 2.....mpla n(atom number) atom1 atom 2.....enterfmolkilllinkmatrpboxpackundo c**? C**?telp cellxl 计算方法在ins中任何地方插入mpla 虚拟平面的原子个数(例如六个原子只有四个可能共平面,即输入4),后面连续输入可能共平面的4个原子,后面在输入其他两个平面外的原子。
例如c1 c2 c3 c4 c5 n1中,c1 c2 c4 c5 共平面mpla 4 c1 c2 c4 c5 c3 n1txt运行xcif选择t两次回车输入文件名.txt选择def回车直到选择q理论加氢在ins中输入HFIX 要加氢的原子保存ins运行XL打开RES拷贝相应的数据到ins中即可。
CHEMICAL DRAW选中画笔点出两个点按ESC点选择键选中画笔鼠标移动至出现小手拖动到其他角度。
单晶结构解析范文单晶结构是固体材料中的一种晶体形态,与此相对的是多晶结构。
在物理学和材料科学领域,对单晶结构的研究具有重要的科学意义和应用价值。
本文将从单晶结构的定义、形成机制以及在科学研究和工业生产中的应用等方面进行解析。
首先,单晶结构是指材料中所有晶体都是同一种密排组织方式的晶体,晶胞参数完全相同,而多晶结构则是指材料中含有多种密排组织方式的晶体。
单晶结构的形成与晶体的生长过程有关。
晶体生长是指固体材料中晶体的逐渐增大和演化的过程。
晶体的生长需要充分的时间和适宜的环境条件。
单晶结构的形成通常需要较长的时间和较高的生长温度。
在恰当的生长条件下,晶体的各向同性增长是单晶的充分条件。
其次,单晶结构在科学研究中具有重要意义。
单晶结构通常具有比多晶材料更高的物理性能,因为晶体结构的完整性更高。
单晶结构对于研究材料的物理性质,例如热膨胀性、热导率、机械性能等具有重要的影响。
通过制备单晶样品,可以准确测量和研究晶体的各向异性和晶格缺陷等特性。
此外,单晶结构的研究对解析材料的微观结构和理解材料的宏观性质也非常重要。
再次,单晶结构在工业生产中也有广泛的应用。
单晶结构的材料通常具有优异的热稳定性和机械性能,因此广泛应用于高温、高压、高性能的工程材料中。
例如,单晶镍基合金被广泛应用于航空发动机中的高温部件,如涡轮叶片、燃烧室衬板等。
单晶结构的金刚石用作高效切削工具,具有极高的硬度和耐磨性。
此外,单晶结构也在电子器件、光学器件等领域得到应用。
最后,对单晶结构进行解析的方法主要包括X射线衍射、电子显微镜等。
X射线衍射是一种广泛应用于单晶结构分析的非破坏性分析方法。
通过测量X射线在晶体中的散射图样,可以确定晶体的晶格参数、晶胞对称性以及晶格缺陷等。
电子显微镜则可以提供更高的空间分辨率,可以用于观察单晶结构中的晶格缺陷和原子构型等细微特征。
综上所述,单晶结构是固体材料中的一种晶体形态,具有独特的结构和性质。
对单晶结构的研究不仅对于科学研究具有重要意义,还在工业生产中得到广泛应用。
单晶体结构解析及材料物性测定方法单晶体结构解析及材料物性测定方法是材料科学领域中的重要研究技术,它对于理解材料的原子结构以及研究材料的性能具有重要意义。
本文将重点介绍单晶体结构解析的原理和常用方法,以及材料物性测定的相关技术。
首先,我们来讨论单晶体结构解析的原理和常用方法。
单晶体结构解析是指通过实验方法确定材料中的晶体结构,即原子或离子在晶格中的排列方式。
这一技术的关键是通过X射线衍射或电子衍射等方法测量晶体表面上的衍射图样,根据衍射的强度和角度信息,经过复杂的计算得到晶体的结构参数。
X射线衍射是最常见的单晶体结构解析方法之一。
该方法利用X射线与晶体的原子间距进行相互作用,通过测量衍射出的X射线在探测器上的衍射图案,可以获得有关晶体结构的信息。
X射线衍射主要包括劳厄衍射和Bragg衍射两种主要模式。
劳厄衍射是指将平面波X射线照射到晶体上,然后测量散射光的强度和角度,从而得到晶体的原子结构。
Bragg衍射则是通过调整X射线和晶体之间的入射角度,使得X射线满足Bragg条件,从而产生最大的衍射峰。
另一种常用的单晶体结构解析方法是电子衍射。
电子衍射是利用电子束与晶体中的原子相互作用产生衍射现象,通过测量衍射图案可以推断晶体的结构。
电子衍射由于其具有更短的波长,因此可以解析出更高分辨率的晶体结构。
该方法在纳米科学研究中得到广泛应用。
单晶体结构解析是物质科学研究的基础,它可以揭示材料的原子级结构信息,对于理解材料的性能和改善材料性能具有重要意义。
通过单晶体结构解析,我们可以了解到材料中原子或离子的排列方式、晶胞参数、晶体对称性等信息,从而可以进一步预测材料的电学、光学、磁学等性质。
接下来,我们将讨论材料物性测定的相关技术。
材料物性测定是指使用实验方法来定量测量材料的物理性质。
材料物性的测定对于材料科学的研究和应用领域都具有重要意义,可以评估材料的性能和应用潜力。
材料的物性可以分为不同的类别,如热学性质、电学性质、光学性质、力学性质等。
单晶结构解析过程
单晶结构解析过程是指通过实验和数据分析来确定晶体中原子的位置、晶格参数和晶体结构的方法。
下面是单晶结构解析的常见步骤:
1. 晶体生长:首先需要获得足够大的单晶样品。
这可以通过各种方法实现,如溶液法、气相法或熔融法。
2. 数据收集:使用X射线衍射技术或中子衍射技术,将单晶样品放置在仪器中,并记录衍射图案。
这些衍射数据包含了不同角度的散射强度和相位信息。
3. 数据处理:对收集到的衍射数据进行处理和分析。
其中一个关键步骤是解析Laue图或斑图,确定晶体的晶系和对称性。
4. 相位问题:由于晶体中的散射信息只包含幅度而没有相位,所以需要采用一些方法来解决相位问题。
常见的方法包括多晶片、重组法、直接法和Patterson法等。
5. 结构求解:根据已解决的相位问题,借助计算机软件或手动计算,进行晶体结构求解。
这个过程包括模型建立、参数优化和误差分析等。
6. 结构修正:对求解得到的初始结构进行修正和调整。
这可能涉及到原子位置的微调、氢原子的添加、电荷密度修正等。
7. 结果验证:最后,通过一系列实验数据和计算方法来验证所得到的晶体结构。
这些包括衍射数据与计算模型之间的比较,键长和键角的合理性,以及物理化学性质的一致性等。
单晶结构解析技巧1. 通常,H原子的处理方法作者要给出:(1)一般通过理论加H,其温度因子为固定值,可通过INS等文件查看(2) 水分子上H原子可通过Fourier syntheses得到(3)检查理论加上的H原子是否正确,主要看H原子的方向。
若不正确则删去再通过Fourier syntheses合成得到(4) 检查H原子的键长、键角、温度因子等参数是否正常。
通过检查分子间或分子内的H键是否合理最易看出H键的合理性(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)(6) 各位来说说方法与心得?2.胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio222_ALERT_3_B Large Non-Solvent H Ueq(max)/Ueq(min) ... 4.97 Ratio342_ALERT_3_B Low Bond Precision on C-C bonds (x 1000) Ang (49)B 级提示当然得重视了。
建议你先把H撤消,精修到C的热椭球不太变形和键长趋正常。
如做不到就要看空间群?衍射点变量比太小?以至追查到原始数据的录取参数和处理等。
这些粗略意见仅供参考,如何?3.在XP中画图时,只有一部分,想长出另外的对称部分。
我是envi完了,然后sgen长出来的,可是和symm显示的对称信息不一样。
比如:我根据envi的结果用sgen O1 4555得到的是O1A而不是O1D,这跟文献中标注的不一样啊,怎么统一呢?很困扰,忘达人指教。
xp里是按顺序编号的,第一个sgen出的的统一为A,依次标号。
你如果想一开始就统一D的话,重新name一下4.高氯酸根怎么精修呀?我用的SHETXL6.1版的,最好告诉我怎么用其中的XSHELL来做,我觉得他好用!Method 1DFIXDfix 1.42 0.02 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Dfix 1.42 0.02 O1 O2 O1 O3 O1 O4 O2 O3O2 O4O3 O4Method 2SADISadi 0.01 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Sadi 0.01 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O45. 晶体的无序是怎么造成的呀,是晶体培养的问题吗?如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5大于0.5没什么的,解完后都在1以下就可以了。
特殊的比较大的在重原子附近也没有关系5.比较确切的定义是单胞中你测定的或你设想的“化学式”的数目。
在分子晶体中,Z 是分子数,在其它各类晶体中则为化学式个数。
例如有机物一般是分子数目,离子晶体像NaCL只好说化学式为4。
Z 的数字决定于你的化学式。
三斜晶系的P-1空间群的Z多为2。
由于双聚等原因如将双聚体写成你的化学式,那么Z就变为1了。
但是就拿这个三斜晶系来说,出现Z为4或6的情况也是可能的。
这时分子形成双聚或者三聚,而你指定的分子式只是个单体罢了。
测定结构初期得到单胞以后,往往希望知道单胞中有几个"分子",如你知道了或提出了化学式,从我们介绍的范氏半径或原字体积即可毛估这个为整数的Z了,不必测密度就知道Z是很有意义的,因分子式有误Z将不会合理,你不愿试一试?6.其实现在水簇或者各种氢键越来越受到重视。
但是给水加氢总是遇到很多麻烦,大家都希望能有一些实际可以用的经验来指导一下大家。
您能给我们一些建议吗?有时候键长不合理(但是又没有其他合适的参峰),键角合理,您说这样如何加氢?我仔细浏览了论坛的一些文章,您能给这些经验做一个评价吗?哪位大侠清楚DFIX和AFIX的用法及格式,请详细指点一下。
Thanks a lot还有,差值加氢的时候如果找不到q,可以用cent产生个点,然后命名为氢吗??把O原子改成C原子,然后再看Q有没有了;如果没有放大Q值到100,不行放大到500个Q值我想你会加上去的!固定H的温度因子为母原子的1.5倍,然后再把C改成O,计算,大功告成我非常关心这两条经验。
麻烦您!从这个贴子了解到大家对H的指定问题非常热中,只好再谈谈我的看法仅供大家参考了。
我曾表白原则上X光衍射不能准确测定H的位置,现在我仍旧这样看而不太懂所谈经验。
即使是低温也很困除非是做低温的中子衍射工作,不过眼下我们没有条件且无法短平快!在网上曾经提出过一些有助于寻找H位置的想法,因为水在晶体中必定与周边形成氢键。
可是如一位网友所说水处在对称中心的位置上时,那么可肯定H呈无序分布无法指定的。
反过来讲如果一个结构报告把H参数都准确列出,我们可以认定这是一篇高水平的研究。
理论加H是基于分子几何构型指定H 的辅助方法,水和甲基等等都不是它可应付得来的。
看来是介绍能量优化理论计算来指定H的时候了,将请国武老师贴出两篇好文章供分享。
有关的计算程序已在论文中列出并可在网上下载,希望这种“理论加氢”方法得以推广。
参考文献1. Z. Anorg. Allg. Chem., 2003, 629: 666-6722. Acta Cryst., 2004, B60: 179-183首先,x-ray衍射是不能准确确定H的真实位置的。
其次,对于N,O等上面的活性氢最好在dmap上找。
再次,H的位置的确定一定要考虑其化学合理性,即H和周围原子形成的氢键的键长键角要合理。
基于上述原则,具体的方法一般是:1。
先在dmap上找到活性氢的位置,如果找不出,就给更大的Q值,然后用dfix固定,在ins文件里编辑。
2。
精修后,检查H的位置是否化学合理。
3。
在xp里面,用himp固定X-H的键长。
4。
在ins文件里,将dfix去掉,同时在新加的氢的前面加上afix 03,并将其温度因子改为-1. ——————————————————————————处理水上H的一个小技巧通常有机骨架上的C和N以及羟基(-OH)上的H都可以利用几何加氢的方法来处理,但是水分子上的氢却不能这样做。
在晶体学数据比较好的情况下,水分子的H原子在残余峰上还是有迹可循的。
通常需要考虑两方面的因素:1)H-O-H的键长和键角(个人认为键长应在0.65-1.15之间,键角在95-115之间);2)氢键,水分子的H应位于能形成合适的氢键位置上,而不是随意的位置。
基于以上两点考虑,在用SHELXTL程序精修时,在主体骨架都确定之后把残余峰的数量改为50,甚至更大(PLAN 50),然后在O周围的残余峰中仔细辨认,把位置合适的残余峰定为H。
从残余峰中得到H原子,键长一般不是理想的键长,而且位置在经修过程可能会发生改变,为了解决这些问题,我们可以这样来做。
在精修以前打开*.ins文件,找到先前确定的H原子,如:加入AFIX 3的指令可以把找到的H固定,同时把H的温度因子固定为父原子的1.2倍。
改为如下:AFIX 3H1 x1 y1 z1 11.0000-1.2000H2 x2 y2 z2 11.0000-1.2000AFIX 0改完保存之后再做精修。
(解决H原子位置变化的问题)H-O的键长,可以在xp界面下用himp指令来解决。
运行xp时,在file之前运行一次HIMP,就可以把H-O调整为理想的0.85A。
这样就基本上可以得到较好的水分子的H原子了。
此外还可以通过WinGX程序包中的CACL-OH来计算水上的H原子。
如果以上两种方法都得不到较好的H原子,建议放弃加氢,因为并不一定每个结构中的水分子的氢都能确定,虽然理论存在可能性。
欢迎大家讨论,共同进步!1.呵呵,刚刚注册,很不错的网站.以后大家可以经常讨论一些结构解析的问题.我有一个结构,游离水的氢通过差值傅里叶定位总出错(温度因子过高),不知道各位大侠有什么高见"把氧原子改为氮原子,并把该氮原子和其他原子连起来,然后理论加氢,把原子名称改回去,把氢原子的参数保留,加入命令AFIX 3H1a .............................H2a................................AFIX 0就可以拉"呵呵,能用差傅里叶峰投H已经是很好的情况了,楼上的法子不太好,那样得到的键角好象有偏差吧。
我们一般是限制键长和键角,固定温度因子,然后就一切OK了2.求助空间群转换教程:其实转换起来也比较容易很多情况下,xprep程序提示的高低不同的空间群CFOM值差别并不是很大,如P1和P-1Cc与C2/c,但是直接按照高的空间群无法显示出分子,或只能看到一部分这时。
可以将空间群降低,先解出绝大部分的结构,加不加氢均可,然后点击platon中的addasym计算空间群,如果提示空间群需要升高,在原文件夹中出现一个platon.res文件,改名为您原来的文件名,用xshell或者xp打开,继续解析即可.高的空间群无法解析的时候,被迫用低空间群解的例子还有一种情况:程序建议采用低空间群,这时用地的空间群可以解出大部分的结构,但是配体的一部分怎么也连不上,grow后也无济于事,这是空间群错误的一个信号,这时我们就可以去升高空间群了.据经验,升高空间群时我们只需要解析出主要骨架的结构就可以了,溶剂分子可以不处理,甚至有时只需把所有的在转换后,经常出现参数溢出的情况,无法继续精修,据经验一般是以下原因造成的:(1) 存在Q峰,在对称性发生变化后,出现混乱)(2) 存在H原子无法抵消造成的混乱以上两种情况提示我们在对称性转换前最好删掉所有H原子和Q峰。
(3)转换后通常会得到FMAP 0的情况,只需将0改为2 就可以了(4)如果带着氢原子去转换,往往会出现一个碳原子上有很多氢的情况,这时建议删掉所有的氢原子,重新加氢另外,需要说明的是:空间群的转换经常会造成R之间的差异较大,这种情况经常出现,需要注意。
In XP mode, the information can be easily obtained from MATR command. For example, when the chain along the (101) direction, Matr 1 0 0 0 0 0 0 0 1, you will find the chain just appeared as a motif. Then you ROTA 1 90, a chain will be observed.晶体结构分析中H 的指定游离的水加氢时,我认为是有可能但危险!所谓理论加氢的根据是几何构型的需要,对于水和甲基等的氢,我们很难说出它的取向,不过水多半形成氢键,这就提供了线索。