单晶结构解析XP作图
- 格式:docx
- 大小:664.03 KB
- 文档页数:9

单晶结构解析XP作图:1、画结构图打开Shelxtl软件→导入res文件→打开XP程序→输入fmol→kill $q→kill $h→envi (中心对称原子)→回车后寻找不同于1555对称操作的代码如2555→sgen 2555→proj(查看结构)→利用操作按钮转动结构找出最佳摆放位置(高度不能大于宽度)→labl 2 500(2 500是默认的大小) →telp 0 -30 0.05 0(后面四个数据分别确定结构的模型和一些参数) →回车回车直到出现Plotfile:(在这里输入名字,这里画结构图,可以统一命名为jiegou) 后回车将会出现命名所有原子的图(根据鼠标位置提醒依次在原子周围左键点击)→draw jiegou→回车后会出现SLPT device[L]:(输入a或者h)再回车输入jiegou,然后回车直到光标不闪位置→出现了xp《图标说明结构图已经画好→quit注:红色字体为结构需要对称操作才能显示一个完整的分子结构所进行的操作。
图片操作解析在这里必须输入命名,否者打不开。
在这里找到res文件的位置2导入res文件后,打开XP操作系统输入fmol后回车3输入kill $h $q4 proj后出现下图所示利用这些按钮旋转得到最佳摆放模式如下图5 按步骤画图二、作堆积图Fmol→matr 1(代表a方向堆积) →pbox 15 15→pack 然后点击按钮sgen/fmol保存(倒数第二个)→proj cell→telp cell →命名duiji 后出现一个命名原子的图框,标出O a b c→命名所有原子当出现这个时表明图已经画好draw duiji→a或h→duiji→→quit1、此步骤可有可无这个键保存标出Oabc即可后按B键保存退出后面是一样的,quit退出程序三、画弱相互作用:氢键、p···π、π···π、M···M等首先要找到氢键的位置,然后再根据对称操作找到氢键画法基本步骤为fmol→kill $q→uniq 原子1 原子2(为氢键的两个原子如uniq C5 N4) →envi N4 3(通过N4对称操作找到C5的操作代码不能为1555如为2555) →sgen 2555→join 3 H5 N4(3代表虚线、注意氢键的因为H5和N4非C5和N4,如错误,可用undo C5 N4来断开这之间的) →pick(杀掉无关的原子:回车键为杀原子,空白键为跳过,/键为保存,注意杀完后不是按/键退出的将没有保存)→telp后面画图步骤与上面一致(只需要标出C5和N4原子)π···π画法对称出两个需要连接的苯环后sgen后→cent/x C5 C6 C7 C8 C9 C10(定义苯环C5C6C7C8C9C10的中心为X1A) →cent/x C15 C16 C17 C18 C19 C20(定义苯环C15C16C17C18C19C20的中心为X1B) →join 3 X1A X1B→proj摆放最佳→pick→telp(画π···π键不需要命名原子telp后命名后出现的命名原子框可以之间按B键退出)以下面画C15-H15A···N3为例经过对称操作后应该为H15C和N3相连此时若不需要再画其他氢键了,可以将两边没有连键的原子杀掉如下图杀完原子后按/键保存退出将显示所杀掉的原子数依次telp只需要命名两个相关的原子即可enter为跳过原子,然后按B键保存退出,或者一直enter到最后也可以保存退出。

用XP程序画单晶结构以及晶胞堆积图一、画单晶结构以及无中断画晶胞堆积图:1.将后缀为res的文件(比如为50710c.res)复制到硬盘根目录下(比如E:盘)2.启动单晶分析软件XP3.XP>>read e:\ 50710c ↵4.XP>>fmol ↵5.XP>> kill $q ↵:删除溶剂分子以及不需要的氢原子等。
6.XP》undo :删除不需要的化学键,原子编号之间用空格间隔开。
如删除硝酸根与金属相连的配位键输入undoLa1 N6 La1 N7。
7.XP>>proj ↵出现画图框,你可以按右上的按钮旋转或显示所有的原子。
将分子结构旋转到适当位置,也就是旋转的尽可能使每个原子都不被挡住。
当旋转到适当位置后按右上角的exit退出8.XP>>telp 0-30 ↵回车至现plorfile:的提示符9.plotfile:mol ↵mol是为你所要画的分子结构图起的名字,你也可以起别的名字。
注意:当你按了回车后会进入画图界面,这时候鼠标已经变成了一个矩形框。
将矩形框移动到显示的原子边上合适位置后点击鼠标左键,就对该原子标注了它的顺序。
鼠标在你标注了第一个原子后会自动移到下一个要标注的原子上或旁边,你仍将矩形框移动到合适位置后按鼠标左键进行标注。
重复以上的操作,直至将全部原子标注完毕。
然后按键盘上的b键保存后自动退出到XP程序的dos操作界面10.XP>>draw mol↵mol还是刚才的文件名11.当你上步操作按完回车后会出现一句话,可能的意思是:将该图形保存成什么格式的文件,有几个选项。
请选择按键盘上的a键(可能是保存成可用acrobat 打开的文件),然后回车12.为你的图形取一个名字,你可以仍用mol13.上步回车后又会出现一句话,意思是要保存为黑白图形还是彩色图形。
输入C 保存成彩色图片。
以上就画好了分子结构图。
你可以在这时候推出XP,即在XP>>提示符后输入quit或exit。


1.通常,H原子的处理方法作者要给出(1)一般通过理论加H,其温度因子为固定值,可通过INS等文件查看(2) 水分子上H原子可通过Fourier syntheses得到(3)检查理论加上的H原子是否正确,主要看H原子的方向。
若不正确则删去再通过Fourier syntheses合成得到(4) 检查H原子的键长、键角、温度因子等参数是否正常。
通过检查分子间或分子内的H键是否合理最易看出H键的合理性(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)(6)各位来说说方法与心得?2.胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio222_ALERT_3_B Large Non-Solvent H Ueq(max)/Ueq(min) ... 4.97 Ratio342_ALERT_3_B Low Bond Precision on C-C bonds (x 1000) Ang (49)B 级提示当然得重视了。
建议你先把H撤消,精修到C的热椭球不太变形和键长趋正常。
如做不到就要看空间群?衍射点变量比太小?以至追查到原始数据的录取参数和处理等。
这些粗略意见仅供参考,如何?3.在XP中画图时,只有一部分,想长出另外的对称部分。
我是envi完了,然后sgen长出来的,可是和symm显示的对称信息不一样。
比如:我根据envi的结果用sgen O1 4555得到的是O1A而不是O1D,这跟文献中标注的不一样啊,怎么统一呢?很困扰,忘达人指教。
xp里是按顺序编号的,第一个sgen出的的统一为A,依次标号。
你如果想一开始就统一D 的话,重新name一下4.高氯酸根怎么精修呀?我用的SHETXL6.1版的,最好告诉我怎么用其中的XSHELL来做,我觉得他好用!Method 1DFIXDfix 1.42 0.02 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Dfix 1.42 0.02 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O4Method 2SADISadi 0.01 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Sadi 0.01 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O45. 晶体的无序是怎么造成的呀,是晶体培养的问题吗?如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5大于0.5没什么的,解完后都在1以下就可以了。

§4 单晶X射线结构分析1.shelxtl.exe程序安装打开bruker\Shelxtl\Disk1\setup.exe程序,安装在C: or D: 根目录下生成C:\SAXI\SXTL\shelxtl.exeSHELXTL主要子程序XPREP:处理由Bruker的XSCANS和SMART系统输出的衍射数据。
该程序是为SHELXTL特别设计的,可用于确定晶体的空间群、转换晶胞参数和晶系、对衍射数据做吸收校正、合并不同颗晶体的衍射数据、对衍射数据进行统计分析。
画出倒易空间图和柏特森截面图、输出其他子程序所需的文件等。
以下段落中,“code”代表化合物的代码。
输入文件:从SAINT+ (CCD) 得到的衍射点数据文件code.raw(已经被还原、尚未做吸收校正的数据),code.hkl和code.p4p文件;或从XSCANS(P4)得到的code.raw,code.p4p,code.psi文件输出文件:用于输入XS/XL子程序、包含分子式和空间群等信息的文件code.ins 以及衍射点强度数据文件code.hkl;记录晶体空间群、晶体外观、衍射数据收集条件以及所使用的有关软件的文件code.pcf;用于XCIF子程序的记录文件code.prpXS:通过直接法或帕特森法计算出试验性的初始结构模型(初始套)。
输入文件:code.ins,code.hkl输出文件:计算结果文件code.res;记录文件code.lstXL:根据初始结构模型(包括原子的种类、位置和原子位移参数)与观察到的衍射强度,对结构模型进行F或 F傅里叶合成计算和最小二乘法精修。
输入文件:code.ins,code.hkl输出文件:code.res,code.lst,以及由code.ins文件中的ACTA指令产生的晶体信息文件code.cif,结构因子(计算和观测值)文件code.fcfXP:检查XS和XL的计算结果、指认原子的种类和标号、观看分子结构模型和晶体堆积图、检查结构模型的“化学合理性”,同时根据发表论文的需要,画出最后结构模型的各种分子结构和晶体堆积图。


单晶结构解析XP作图:1、画结构图打开Shelxtl软件→导入res文件→打开XP程序→输入fmol→kill $q→kill $h→envi (中心对称原子)→回车后寻找不同于1555对称操作的代码如2555→sgen 2555→proj(查看结构)→利用操作按钮转动结构找出最佳摆放位置(高度不能大于宽度)→labl 2 500(2 500是默认的大小) →telp 0 -30 0.05 0(后面四个数据分别确定结构的模型和一些参数) →回车回车直到出现Plotfile:(在这里输入名字,这里画结构图,可以统一命名为jiegou) 后回车将会出现命名所有原子的图(根据鼠标位置提醒依次在原子周围左键点击)→draw jiegou→回车后会出现SLPT device[L]:(输入a或者h)再回车输入jiegou,然后回车直到光标不闪位置→出现了xp《图标说明结构图已经画好→quit注:红色字体为结构需要对称操作才能显示一个完整的分子结构所进行的操作。
图片操作解析在这里必须输入命名,否者打不开。
在这里找到res文件的位置2导入res文件后,打开XP操作系统输入fmol后回车3输入kill $h $q4 proj后出现下图所示利用这些按钮旋转得到最佳摆放模式如下图5 按步骤画图二、作堆积图Fmol→matr 1(代表a方向堆积) →pbox 15 15→pack 然后点击按钮sgen/fmol保存(倒数第二个)→proj cell→telp 命名所有原子当出现这个时表明图已经画好cell→命名duiji 后出现一个命名原子的图框,标出O a b c→draw duiji→a或h→duiji→→quit1、此步骤可有可无这个键保存标出Oabc即可后按B键保存退出后面是一样的,quit退出程序三、画弱相互作用:氢键、p···π、π···π、M···M等首先要找到氢键的位置,然后再根据对称操作找到氢键画法基本步骤为fmol→kill $q→uniq 原子1 原子2(为氢键的两个原子如uniq C5 N4) →envi N4 3(通过N4对称操作找到C5的操作代码不能为1555如为2555) →sgen 2555→join 3 H5 N4(3代表虚线、注意氢键的因为H5和N4非C5和N4,如错误,可用undo C5 N4来断开这之间的) →pick(杀掉无关的原子:回车键为杀原子,空白键为跳过,/键为保存,注意杀完后不是按/键退出的将没有保存)→telp后面画图步骤与上面一致(只需要标出C5和N4原子)π···π画法对称出两个需要连接的苯环后sgen后→cent/x C5 C6 C7 C8 C9 C10(定义苯环C5C6C7C8C9C10的中心为X1A) →cent/x C15 C16 C17 C18 C19 C20(定义苯环C15C16C17C18C19C20的中心为X1B) →join 3 X1A X1B →proj摆放最佳→pick→telp(画π···π键不需要命名原子telp后命名后出现的命名原子框可以之间按B键退出)以下面画C15-H15A···N3为例经过对称操作后应该为H15C和N3相连此时若不需要再画其他氢键了,可以将两边没有连键的原子杀掉如下图杀完原子后按/键保存退出将显示所杀掉的原子数依次telp只需要命名两个相关的原子即可enter为跳过原子,然后按B键保存退出,或者一直enter到最后也可以保存退出。


晶体精修解析后的画图方法2009-06-29 16:31查看晶体图运行时要求存在两个文件.hkl;.ins;F:\dir leeF:\cd leeF:\lee >fmol less $cF:\lee >fmol less c101 to c112 c201 to c212F:\lee >edit p.cif返回上一层结构:原先:F:\lee >输入cd..变为F:\lee>cd.. 按回车键得到:F:\ >ClO4为球状基团,晶体解析为无序结构。
晶体结构精修解析到可以发文章才可以。
精修好画图:原先:C:\ Documents and Settings\Administrator\输入F: (F指晶体数据储存在F盘中)得到F: \输入cd lee得到F: \ lee输入xp p得到xp>>输入proj按回车键,查看并调整图的角度至最好,在键盘上按Esc选择在某一个最好的角度并且不要退出或者再改变角度,这用于画同一个角度下只显示部分或全部原子的不同图。
得到:xp>>输入labl 1 250得到:xp>> labl 1 250按回车键得到:Labels switched on for 22 of 22 current atoms,code=1,size=250. (22指原子数目,code=1指以第一种方式为原子添加标签,250指字体大小,可以从1到500中任选一个数字,一般选择250,300,350)再输入telp 30 (30指椭球概率,默认选择30)得到:xp>> telp 30按回车键得到:Plotfile:输入:a (a指你想用a来命名产生的.plt文件,.plt文件作用是产生.ps文件) 按回车键得到:左右两个几乎相同的图形,开始给原子加标签。
界面的右下角有将要加标签的原子的说明,需要加标签的原子移动鼠标于合适位置(不遮住原子和键,又靠近需要加标签的原子)点击鼠标左键,点击鼠标左键后不要随意移动鼠标,否则你不知道下一个原子是哪一个,不需要加标签的原子直接按回车键。

椭圆球图形的画法:1.将后缀为res的文件(比如为50710c.res)复制到硬盘根目录下(比如E:盘)2.启动单晶分析软件XP3.XP>>read e:\ 50710c ↵4.XP>>fmol ↵5.XP>> kill $q ↵6.XP>>proj ↵出现画图框,你可以按右上的按钮旋转或显示所有的原子。
将分子结构旋转到适当位置,也就是旋转的尽可能使每个原子都不被挡住。
当旋转到适当位置后按右上角的exit退出7.XP>>labl 1 450 ↵8.XP>>telp 0-30 ↵回车至现plorfile:的提示符9.plotfile:mol ↵mol是为你所要画的分子结构图起的名字,你也可以起别的名字。
注意:当你按了回车后会进入画图界面,这时候鼠标已经变成了一个矩形框。
将矩形框移动到显示的原子边上合适位置后点击鼠标左键,就对该原子标注了它的顺序。
鼠标在你标注了第一个原子后会自动移到下一个要标注的原子上或旁边,你仍将矩形框移动到合适位置后按鼠标左键进行标注。
重复以上的操作,直至将全部原子标注完毕。
然后按键盘上的b键保存后自动退出到XP程序的dos操作界面10.XP>>draw mol↵mol还是刚才的文件名11.当你上步操作按完回车后会出现一句话,可能的意思是:将该图形保存成什么格式的文件,有几个选项。
请选择按键盘上的a键(可能是保存成可用acrobat打开的文件),然后回车12.为你的图形取一个名字,你可以仍用mol13.上步回车后又会出现一句话,意思是要保存为黑白图形还是彩色图形。
按回车即保存成黑白图形,请按回车。
以上就画好了分子结构图。
你可以在这时候推出XP,即在XP>>提示符后输入quit或exit。
你也可以继续画堆积图。
14.XP>>cell ↵回车后会出现5个数字,前3个数字依次表示的是a、b、c轴。

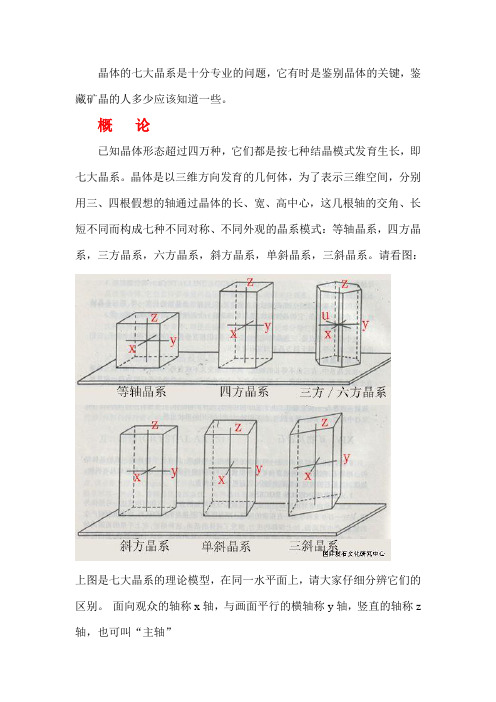
晶体的七大晶系是十分专业的问题,它有时是鉴别晶体的关键,鉴藏矿晶的人多少应该知道一些。
概论已知晶体形态超过四万种,它们都是按七种结晶模式发育生长,即七大晶系。
晶体是以三维方向发育的几何体,为了表示三维空间,分别用三、四根假想的轴通过晶体的长、宽、高中心,这几根轴的交角、长短不同而构成七种不同对称、不同外观的晶系模式:等轴晶系,四方晶系,三方晶系,六方晶系,斜方晶系,单斜晶系,三斜晶系。
请看图:上图是七大晶系的理论模型,在同一水平面上,请大家仔细分辨它们的区别。
面向观众的轴称x轴,与画面平行的横轴称y轴,竖直的轴称z 轴,也可叫“主轴”一,等轴晶系简介等轴晶系的三个轴长度一样,且相互垂直,对称性最强。
这个晶系的晶体通俗地说就是方块状、几何球状,从不同的角度看高低宽窄差不多。
如正方体、八面体、四面体、菱形十二面体等,它们的相对晶面和相邻晶面都相似,这种晶体的横截面和竖截面一样。
此晶系的矿物有黄铁矿、萤石、闪锌矿、石榴石,方铅矿等。
请看这种晶系的几种常见晶体的理论形态:等轴晶系的三个晶轴(x轴y轴z轴)一样长,互相垂直。
常见的等轴晶系的晶体模型图金刚石晶体八面体和立方体的聚形的方铅矿黄铁矿二,四方晶系简介四方晶系的三个晶轴相互垂直,其中两个水平轴(x轴、y轴)长度一样,但z轴的长度可长可短。
通俗地说,四方晶系的晶体大都是四棱的柱状体,(晶体横截面为正方形,但有时四个角会发育成小柱面,称“复四方”),有的是长柱体,有的是短柱体。
再,四方晶系四个柱面是对称的,即相邻和相对的柱面都一样,但和顶端不对称(不同形);所有主晶面交角都是九十度交角。
请看模型图:四方晶系的晶体如果z轴发育,它就是长柱状甚至针状;如果两个横轴(x 、y)发育大于竖轴z轴,那么该晶体就是四方板状,最有代表性的就是钼铅矿。
请看常见的一些四方晶系的晶体模型:这个晶系常见的矿物有锡石、鱼眼石、白钨矿、符山石、钼铅矿等。
请看实物图片:符山石的晶体锡石的长柱状晶体(顶端另有斜生的小晶体)。

晶体结构查看和修改软件XP应用指南1.进入界面直接点击XP.exe的应用程序图标。
或者从命令提示符进入。
2.如何打开一张RES图?(1)首先确定RES扩展名的文件的位置及其文件名。
然后将其拷到XP所在的文件夹中。
例如:在我的文档中有一个08122m . res的文件(2)在XP的界面中如下操作:XP》read 08122mXP》fmol按enter键后,会出现一系列数据标示。
按enter使屏幕滚动,提示符重新出现。
XP》mpln/n (选择项)(分子沿对称轴方向定向)屏幕出现x . y . z 轴坐标。
XP》proj即可进入08122m的XRD结构图,右上角有一系列命令3.如何只显示部分图形?方法一:XP》pick屏幕出现一张结构全图,并自动扫描各键。
确定拟保留和除去的键及原子,然后当扫描到某个键时,按“空格键”保留该键,按“enter”键删除该键。
依次操作。
依次操作直至满足要求。
①当误删时,按“backspace”即可恢复上步操作。
②处理完毕,按“ESC”返回至XP》____ 状态。
但保留处理。
③处理完毕,按“?/”键即可将处理后的图保留下来。
④当扫描到某个原子时,可键入元素符号和数字来改变原子的编号。
方法二:XP》pick $C则光标自动扫描所有C原子。
按“空格键”或“enter”键来决定是逐个保留还是删除。
方法三:XP》kill P1此法需要在已知拟删原子的坐标符合之后才能运用。
否则很容易误删。
上面的命令可删除结构图上的P1原子,然后给出“一个原子被删”的提示后直接回到XP》_____.以上三个方法均可用通配符“?”或“*”,C1 to C64.如何进行晶胞堆积?(1)按X,Y或Z轴方向堆积:XP》matr 1 (按X方向堆积)XP》matr 屏幕会给出一系列参数XP》pack 屏幕会给出在一定长、宽范围沿X轴的晶胞堆积图,用红蓝两色给出。
若戴上一幅红蓝镜片的眼镜,则可看到立体图。
如果要贮存,在退出时选择右上方“SGEN/FMOL”命令,那么接下来用PROJ命令打开时,即为该图,而不是以前的单晶。



单晶结构解析XP作图:
1、画结构图
打开Shelxtl软件→导入res文件→打开XP程序→输入fmol→kill $q→kill $h→envi (中心对称原子)→回车后寻找不同于1555对称操作的代码如2555→sgen 2555→proj(查看结构)→利用操作按钮转动结构找出最佳摆放位置(高度不能大于宽度)→labl 2 500(2 500是默认的大小) →telp 0 -30 0.05 0(后面四个数据分别确定结构的模型和一些参数) →回车回车直到出现Plotfile:(在这里输入名字,这里画结构图,可以统一命名为jiegou) 后回车将会出现命名所有原子的图(根据鼠标位置提醒依次在原子周围左键点击)→draw jiegou→回车后会出现SLPT device[L]:(输入a或者h)再回车输入jiegou,然后回车直到光标不闪位置→出现了xp《图标说明结构图已经画好→quit
注:红色字体为结构需要对称操作才能显示一个完整的分子结构所进行的操作。
图片操作解析
在这里找到res
文件的位置
2导入res 文件后,打开XP 操作系统输入fmol 后回车
3输入kill $h $q
4 proj 后出现下图所示
5 按步骤画图
命名所有原子
二、作堆积图
Fmol→matr 1(代表a方向堆积) →pbox 15 15→pack 然后点
击按钮sgen/fmol保存(倒数第二个)→proj cell→telp cell
→命名duiji 后出现一个命名原子的图框,标出O a b c→draw duiji→a或h→duiji→→quit
1、
当出现这个时表
明图已经画好
这个键
保存
此步骤可有可无
标出Oabc即可后按B键保存退出
后面是一样的,quit退出程序
三、画弱相互作用:氢键、p···π、π···π、M···M等
首先要找到氢键的位置,然后再根据对称操作找到
氢键画法
基本步骤为fmol→kill $q→uniq 原子1 原子2(为氢键链接的两个原子如uniq C5 N4) →envi N4 3(通过N4对称操作找到C5的操作代码不能为1555如为2555) →sgen 2555→join 3 H5 N4(3代表虚线、注意氢键链接的因为H5和N4非C5和N4,如链接错误,可用undo C5 N4来断开这之间的链接) →pick (杀掉无关的原子:回车键为杀原子,空白键为跳过,/键为保存,注意杀完后不是按/键退出的将没有保存)→telp后面画图步骤与上面一致(只需要标出C5和N4原子)
π···π画法
对称出两个需要连接的苯环后sgen后→cent/x C5 C6 C7 C8 C9 C10(定义苯环C5C6C7C8C9C10的中心为X1A) →cent/x C15 C16 C17 C18 C19 C20(定义苯环C15C16C17C18C19C20的中心为X1B) →join 3 X1A X1B→proj摆放最佳→pick→telp(画π···π键不需要命名原子telp后命名后出现的命名原子框可以之间按B键退出)
以下面画C15-H15A···N3为例
此时若不需要再画其他氢键了,可以将两边没有连键的原子杀掉如下图
杀完原子后按/键保存退出
将显示所杀掉的原子数
依次telp
只需要命名两个链接相关的原子即可enter为跳过原子,然后按B键保存退出,或者一直enter到最后也可以保存退出。
然后draw与上面一致。
在同一个图中画多个氢键相对复杂一些,按步骤进行,画完一个连接一个,保留需要画氢键所需要对称的那个原子所在的结构,然后杀掉其余不需要用到的,然后再画另外一个。
(一定要先杀掉不需要用到的结构,否者对称操作后会很混乱)
其余弱相互作用画法类似,p···π用O原子跟苯环的中心相连。