实例图解简并引物设计
- 格式:ppt
- 大小:3.55 MB
- 文档页数:16

干货:引物设计有妙招,有图有真相PCR现在已经成为分子实验室的标配,无论是基因检测、基因定量、分子克隆、组装、突变、测序等等,都离不开PCR技术。
在PCR 过程中,引物设计是非常重要的一环,特别是qPCR的引物设计,决定了我们扩增效率是否达标和扩增产物是否特异。
今天小编为大家举个例子,设计一对qPCR引物。
qPCR引物设计,通常要留意以下几点:GC含量50%-60%Tm值50℃-65℃上下游引物尽量接近引物末端最好是G或C避开二级结构复杂区域引物尽量在目的基因的3’ 端引物尽量跨越内含子目的基因是否有不同的剪切变体……这么多要注意的怎么办?!Primer-blast相信很多小伙伴们都用过,只要输入序列设置条件就可以设计得到理想的引物序列了。
但是很多情况下,我们的要求往往不仅于此。
下面,我们以人的EGFP (Epidermal growth factor receptor,表皮生长受体因子)基因为例,设计一对可以同时扩增其不同剪切变体的qPCR引物!1. 查询基因结构之前小编已经为大家介绍了如何利用NCBI查询基因结构了(错过了可以戳这里),首先查询Homo EGFP的基因结构:2. 确定设计引物位置可以看到,这个基因有多种不同的转录本,而前8个外显子区域是完全一致的(绿色小竖线)。
我们的目的是将不同的转录本都扩增出来,那将反向引物设在第8个外显子之前就可以实现了。
3. 找到mRNA的序列号,进入GenBank在该页面里继续往下拉,找到mRNA和蛋白质的GenBank序列号。
点击mRNA的序列号,进入GenBank。
借助Highlight Sequence Features,我们可以快速确定第8个外显子的位置。
4. 确定下游引物设计位置点击Highlight Sequence Features后,浏览器的下方就会出现选项,选择查看Exon,找到第8个外显子,这样就能快速定位了。
我们确定了第8个外显子到下游至1236bp为止。
如何根据要求自己设计PCR引物1PCR引物设计课堂笔记○PCR这个名词大家都不陌生,但实际操作时我们常说的引物设计到底是怎么回事呢?今天我就来给大家用实例演示一下哈。
首先,我们要知道引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。
引物设计是PCR的关键,附上PCR的基本流程图:○引物设计的原则:1.引物长度:一般为15-30bp,常用的是18-27bp,但不能大于38,因为过长会导致其延伸温度大于74℃,即Taq酶的最适温度。
2.引物的特异性:引物与非特异扩增序列的同源性不要超过70%或有连续8个互补碱基同源。
3.序列Tm值:引物的Tm值一般控制在55-60度, 尽可能保证上下游引物的Tm值一致,一般不超过2度。
退火温度=4×(G+C)+2×(A+T)-(5~8)4.G+C含量:有效引物中(G+C)的比例为40-60%,过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
5. 引物的3′端:引物的延伸是从3′端开始的,不能进行任何修饰;引物3’端的碱基一般不用A,因为A在错误引发位点的引发效率相对比较高;引物间3’端的互补、二聚体或发夹结构也可能导致PCR反应失败6.引物的5′端:引物的5′端限定着PCR产物的长度,它对扩增特异性影响不大。
因此,可以被修饰而不影响扩增的特异性。
引物5′端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入突变位点、插入与缺失突变序列和引入一启动子序列等。
下面以实例操作演示一下加酶切位点时如何自己设计引物:2用绿色荧光蛋白(GFP)标记蛋白NR1○简单点说,就是现在我们要把Plasmid 2中GFP基因片段添加到Plasmid 1中的NR1基因片段上,但是Plasmid 2中GFP基因片段本身并没有BamHⅠ这个酶切位点,也就说我们要在引物设计中人为地把BamHⅠ这个酶切位点的序列添加给GFP基因片段,这样PCR后得到的GFP基因片段就可以通过BamHⅠ这个酶切位点进入到Plasmid 1中,然后绿色荧光蛋白(GFP)就可以来标记蛋白NR1,达到我们之后实验中来观察蛋白NR1的目的,示意图见下。



引物设计知识点归纳图解引物设计是分子生物学和遗传学等研究领域中的重要工具,它在PCR扩增、基因克隆、基因表达分析等方面发挥着至关重要的作用。
在引物设计过程中,需要考虑引物的长度、碱基组成、熔解温度等因素,以确保引物的特异性和稳定性。
本文将围绕引物设计的基本原理、常用工具和实践技巧展开讨论,并通过图解的方式进行归纳总结。
一、引物设计的基本原理引物设计的目标是选择具有高特异性和稳定性的引物,以确保在PCR扩增等实验中的准确和可靠性。
其基本原理包括:1. 特异性:引物应与目标DNA序列的特定区域完全匹配,以避免非特异扩增。
2. 稳定性:引物应具有适当的长度和碱基组成,以确保引物在反应条件下的稳定性。
3. 熔解温度:引物的熔解温度应接近PCR扩增的反应温度,以保证引物的特异性。
二、引物设计的常用工具引物设计可以借助多种在线工具和软件来完成,常用工具包括:1. Primer3:一种广泛应用的引物设计工具,可以根据给定的参数自动设计引物,并进行热力学分析。
2. NCBI Primer-BLAST:结合NCBI数据库和BLAST算法的引物设计工具,用于检验引物的特异性。
3. OligoAnalyzer:根据序列特性进行引物设计和分析的在线工具,可以计算引物的熔解温度和配对特性。
三、引物设计的实践技巧在实践过程中,为了提高引物设计的准确性和效率,可以考虑以下技巧:1. 引物长度:引物的长度通常为18-25个碱基对,较短的引物具有更高的特异性。
2. GC含量:引物的GC含量应在40-60%之间,高GC含量可以增强引物的熔解温度和特异性。
3. 引物间配对:引物之间或引物与模板之间的配对应避免,以防止引物间的二聚体形成或引物与模板的非特异结合。
4. 引物位置:引物应位于目标序列的特异区域,避免引物与非目标序列的交叉反应。
5. 引物设计的复杂性:复杂的引物设计场景,如多组引物设计、引物与探针联合设计等,可以借助专业软件进行。

简并引物设计过程及原则简并引物常用于从已知蛋白到相关核酸分子的研究及用于一组引物扩增一类分子。
简并引物设计过程(1)利用NCBI搜索不同物种中同一目的基因的蛋白质或cDNA编码的氨基酸序列因为密码子的关系,不同的核苷酸序列可能表达的氨基酸序列是相同的,所以氨基酸序列才是真正保守的。
首先利用NCBI的Entrez检索系统,查找到一条相关序列即可。
随后利用这一序列使用BLASTP(通过蛋白查蛋白),在整个NR数据库中查找与之相似的氨基酸序列。
(2)对所有的序列进行多序列比对将搜索到的同一基因的不同氨基酸序列进行多序列比对,可选工具有Clustal W/X, 也可在线分析。
所有序列的共有部分将会显示出来。
“*”表示保守,“:”表示次保守。
(3)确定合适的保守区域设计简并引物至少需要上下游各有一个保守区域,且两个保守区域相距50~400个氨基酸残基为宜,使得PCR产物在150~1200bp之间,最重要的是每一个保守区域至少有6个氨基酸的保守区,因为每条引物至少18bp左右。
若比对结果保守性不是很强很可能找不到6个氨基酸序列的保守区,这时可以根据物种的亲缘关系,选择亲缘关系近的物种进行二次比对,若保守性仍达不到要求,则需进行三次比对,总之,究竟要选多少序列来比对,要根据前一次的结果反复调整。
最终目的就是有两个6个氨基酸且两者间距离合适的保守区域。
(4)利用软件设计引物当得到保守区域后,就可以利用专业的软件来设计引物了,其中Primer 5.0 支持简并引物的设计,将参与多序列比对的序列中的任一条导入Primer 5.0 中,将其翻译成核苷酸序列,该序列群可用一条有简并性的核苷酸链来表示(其中R=A/G,Y=C/T,M=A/C, K=G/T, S=C/G, W=A/C/T, B=C/G/T,V=A/C/G, D=A/G/T, N=A/C/G/T, 该具有简并性的核苷酸链必然包含上一步中找到的氨基酸保守区域的对应部分,在Primer 5.0 中修改参数,令其在两个距离合适的保守的nt 区域内寻找引物对,总之要保证上下游引物都落在该简并链的保守区域内,结果会有数对,分数越高越好。
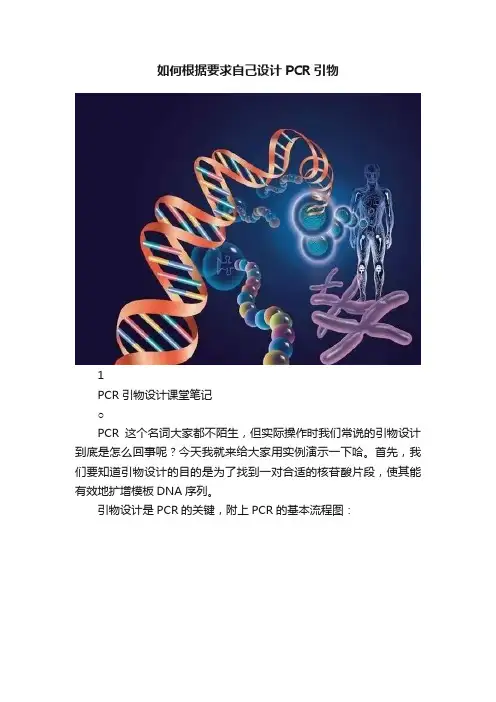
Primer5.0 目的基因序列引物设计
GA TTtCTTGGCTTtATA TA TCTTGT GGAaAGGaCGAAACACCGTGCTCGCTTCGGCAGCAC ATATACTAGTCGACGGGTCTAGACAA TGA TGCTGGGTAA TGACACCAAGCTGGGACTG GTACAGAAAGTCAGAGAACACTTACAGAACGGCA TCTAGACAATGA TGCTGGGTAATA CACTTACAGAACGGCA TCTAGA TGCCGTTCTGTAAGTGTTTGTTGAATGAATGAGTGTT GAACAAACTGCTAAGGTATCTTT ACAAGGTAG
从中扩增出目的基因片段(绿色的部分):
首先:将绿色部分复制到primer5.0引物设计软件中,ctrl+v用鼠标右键不管用
.选择As is后,惦记OK出现:
然后惦记,出现
图中右上角标记,其中惦记S合成的是上游引物,A是下游引物,点击S后出现,然后点击出现
这些就是上游引物,选中后ctrl+c复制到引物合成单中,发给公司,下游引物:回到
点击A出现
用前后调节框,如下,将序列向右移动移到最后,得到下图
注意显示的是3—5,ctrl+c—ctrl+v后悔发现序列变成5---3了,这是软件的好处,因为公司引物合成就是从5—3的
其实上下游引物只是相对的,将得到的引物序列填表后发给公司就好了。
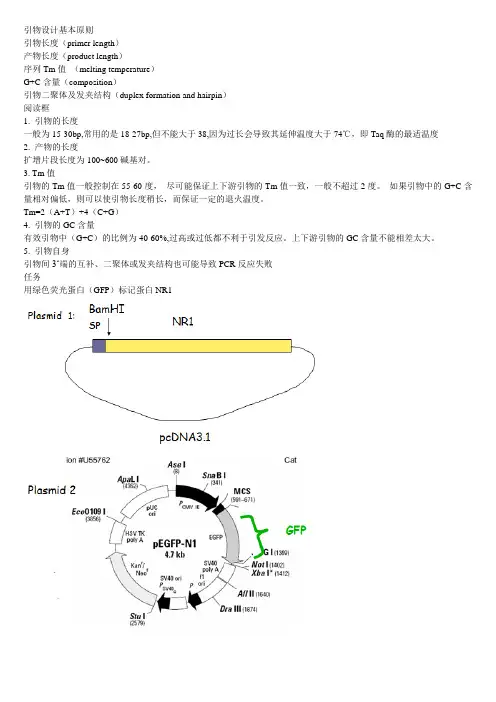
引物设计基本原则引物长度(primer length)产物长度(product length)序列Tm值(melting temperature)G+C含量(composition)引物二聚体及发夹结构(duplex formation and hairpin)阅读框1. 引物的长度一般为15-30bp,常用的是18-27bp,但不能大于38,因为过长会导致其延伸温度大于74℃,即Taq酶的最适温度2. 产物的长度扩增片段长度为100~600碱基对。
3. Tm值引物的Tm值一般控制在55-60度,尽可能保证上下游引物的Tm值一致,一般不超过2度。
如果引物中的G+C含量相对偏低,则可以使引物长度稍长,而保证一定的退火温度。
Tm=2(A+T)+4(C+G)4. 引物的GC含量有效引物中(G+C)的比例为40-60%,过高或过低都不利于引发反应。
上下游引物的GC含量不能相差太大。
5. 引物自身引物间3‘端的互补、二聚体或发夹结构也可能导致PCR反应失败任务用绿色荧光蛋白(GFP)标记蛋白NR1引物要求PCR扩增GFPGFP两边添加BamHI酶切位点保证NR1的阅读框不改变第一步:扩增GFP基本序列第二步:GC比值;Tm值第三步:酶切位点第四步:阅读框第五步保护序列Primer1: 5' GCGGggatccTATGGTGAGCAAGGGCGAGGA Primer2: 5' GCGCggatccctCTTGTACAGCTCGTCCA TGCC记得当初写本科论文,感到不知道讨论什么问题好。
愣是写了一大段的PCR条件摸索的讨论。
后来PCR成为实验最基本的一步了,但是发现在PCR中还是有许多需要注意的地方。
PCR的第一步就是引物设计了。
引物设计需要注意的地方很多,在大多数情况下,我们都是在知道已知模板序列时进行PCR扩增的。
在某些情况比如构建文库的时候也会在不知道模板序列的情况下进行设计。
这个时候随机核苷酸序列就与模板不是完全匹配。

简并引物设计PCR(聚合酶链反应)是一种基础而重要的技术,在生物学、医学、检测等领域都有着广泛的应用。
然而,PCR技术在实际应用过程中,常常会遇到一些问题,例如引物设计不合理、多重特异性等。
简并引物设计便是为了解决这些问题而产生的技术。
本文将介绍简并引物设计的原理、方法、优缺点及应用。
一、简并引物设计的原理简并引物是一种含有多个碱基的引物,其中每个碱基的位置都可以存在多种可能性。
简并引物的设计原理是根据目标序列的多态性,将可能出现的碱基变异情况考虑在内,从而设计出能够特异性扩增目标序列的引物。
二、简并引物设计的方法简并引物设计可以通过人工设计或计算机辅助设计完成。
1. 人工设计人工设计简并引物需要根据目标序列的多态性情况,逐个考虑每个碱基的变异情况,然后选择合适的碱基组合作为简并引物的设计。
例如,对于一段序列“ATGCTAGC”,在第三个碱基位置上存在两种可能性“A”和“T”,在第七个碱基位置上存在三种可能性“A”、“C”和“G”,则可以设计出两个简并引物:ATGCTAGC和ATGCTAGN。
2. 计算机辅助设计计算机辅助设计可以通过在线工具或软件完成,例如Primer3、Primer-BLAST等。
这些工具可以根据用户输入的目标序列信息和扩增条件,自动生成合适的简并引物设计方案。
三、简并引物设计的优缺点简并引物设计相比于传统引物设计有以下优点:1. 提高特异性简并引物设计可以考虑到目标序列的多态性,从而提高扩增的特异性。
特别是对于高度变异的序列,简并引物设计可以有效地避免非特异性扩增。
2. 减少设计时间传统引物设计需要逐个考虑每个碱基的变异情况,而简并引物设计可以通过在线工具或软件自动生成设计方案,大大减少了设计时间。
3. 降低成本简并引物设计可以减少试验重复次数和材料浪费,从而降低实验成本。
简并引物设计的缺点包括:1. 扩增效率可能降低由于简并引物设计需要考虑目标序列的多态性,因此引物长度可能会增加,扩增效率可能会降低。


PCR和简并引物设计PCR(聚合酶链反应)是一种广泛应用于分子生物学领域的技术,它可以通过体外复制DNA片段,从而能够快速、高效地扩增目标DNA序列。
PCR的成功扩增依赖于精确的引物设计,其中简并引物设计是其中一种常用的方法。
本文将对PCR和简并引物设计进行详细的介绍。
PCR的原理和步骤可以概括为三个主要步骤:变性、退火和延伸。
在变性步骤中,PCR反应体系中的DNA双链被加热,使其变性成单链。
接下来,在退火步骤中,引物与单链DNA互相结合,形成DNA引物-模板DNA 复合物。
最后,在延伸步骤中,通过DNA聚合酶的作用,从引物的3'端开始合成新的DNA链。
PCR反应可以通过反复进行这三个步骤来进行多轮扩增,从而快速地产生大量目标DNA片段。
引物在PCR反应中起着至关重要的作用,引物的设计需要满足以下几个要求。
首先,引物的序列应与目标DNA序列互补,并且引物之间没有序列重复。
其次,引物的长度应适当,通常为18-30个碱基对。
引物的长度过长或过短都会影响扩增效率。
最后,引物的GC含量应在40-60%之间,这样可以提高引物与模板DNA的互补性。
相对于常规引物,简并引物设计是一种更加灵活和高效的方法。
简并引物设计的基本思想是在引物的特定位置上引入简并碱基(简并位点)。
简并碱基是可以用多种碱基取代的碱基,它可以与多种目标序列互补,从而提高引物与目标序列的互补性。
简并引物的设计通常需要考虑以下几个因素。
首先,需要确定哪些位置引入简并位点。
一般来说,简并位点通常选择在引物的3'端或其周围,因为3'端是引物与模板DNA结合的起始点,而简并位点可以提高引物的互补性。
其次,需要确定哪些碱基可以在简并位点上使用。
简并碱基通常是由两个或多个非互补的碱基组成,例如,简并位点可以是R(代表A或G)、Y(代表C或T)或M(代表A或C)等。
选择简并碱基时需要考虑目标DNA序列的GC含量和序列特点,以确保引物与目标序列的互补性。
PCR和简并引物设计PCR(聚合酶链反应)是一种在分子生物学和遗传学研究中广泛使用的技术。
它通过扩增DNA特定区域的序列,从而产生大量的DNA复制体。
PCR的核心组成部分是引物(primers),它们是能与待扩增序列的起始点匹配的短DNA片段。
对于PCR的成功与否,引物的设计起到了关键作用。
本文将重点讨论PCR和简并引物设计的原理和方法。
PCR的步骤包括解旋(denaturing)、退火(annealing)和扩增(extension)。
在解旋阶段,样品中的DNA被加热至高温,使其双链结构解开,生成两根单链的DNA模板。
然后在退火阶段,温度被降低,使引物能够与模板上的特定序列进行互补配对,从而定位引物的位置。
最后,在扩增阶段,通过DNA聚合酶的作用,引物延伸成为新的DNA链,产生大量的特定序列。
引物的设计是PCR的关键步骤之一、它们需要满足以下几个条件:合适的长度、特异性、错配最少、避免形成二聚体或自身扩增以及合适的Tm(退火温度)。
首先,引物应该在15-30个碱基长的范围内,通常为18-25个碱基。
太短的引物可能与多个位点杂交,而太长的引物可能会降低特异性和扩增效率。
其次,引物应该是特异性的,即只与待扩增序列的特定区域互补配对。
为了确保特异性,通常需要使用序列比对软件来挑选最好的引物。
比对软件可以帮助我们检查引物是否与其他地方的基因组序列有互补性。
引物的错配最少,也就是引物与待扩增序列最好是完全互补配对。
因为即使有一个碱基错配,也可能导致错误的扩增产物的形成。
可以通过比对引物和目标序列的碱基来评估匹配的情况,以确定引物设计是否合理。
引物应该避免形成二聚体或自身扩增。
二聚体是指引物之间通过互补配对形成的二级结构。
如果引物形成二聚体,它们会被聚合酶识别为模板,导致错误的扩增产物生成。
自身扩增是指引物上的5'端和3'端通过互补配对形成环状结构。
自身扩增也会引起非特异性扩增。
为了避免这些情况的发生,引物的序列应该被合理设计。
在专门的引物设计软件中,“Oligo”是最著名的。
它的使用并不十分复杂,但初学者容易被其复杂的图表吓倒。
Oligo 5.0的初始界面是两个图:Tm图和ΔG图;Oligo 6.0的界面更复杂,出现三个图,加了个Frq图。
“Oligo”的功能比“Premier”还要单一,就是引物设计。
但它的引物分析功能如此强大以至于能风靡全世界。
oligo的下载和安装我就不多说了,打开oligo相信也无需多讲。
打开oligo的页面如下:单击file菜单再点open或点击“打开”快捷图标或者用快捷键“CTrl+O”可打开下面的窗口:在打开的OPEN窗口内选择FreqSeq再点“打开”:选择drosfr或者其它一个文件点击“打开”:出现以下窗口,点击“window”再点击“Tile”:出现以下窗口,图中显示的三个指标分别为Tm、ΔG和Frq,其中Frq是6.0版本的新功能,为邻近6至7个碱基组成的亚单位在一个指定数据库文件中的出现频率。
该频率高则可增加错误引发的可能性。
因为分析要涉及多个指标,起动窗口的cascade排列方式不太方便,可从windows菜单改为tile方式。
如果觉得太拥挤,可去掉一个指标,如Frq,这样界面的结构同于Oligo 5.0,只是显示更清楚了:∆G值反映了序列与模板的结合强度,最好引物的∆G值在5'端和中间值比较高,而在3'端相对低(如图)。
Tm值曲线以选取72℃附近为佳,5'到3'的下降形状也有利于引物引发聚合反应。
Frq曲线为“Oligo 6”新引进的一个指标,揭示了序列片段存在的重复机率大小。
选取引物时,宜选用3'端Frq值相对较低的片段:再点击Search再点“Fo'r Primers and probes”或使用快捷键F3:出现以下窗口,点“OK”就OK了。
当然你也可以点击“Prameters”和“SearchRange”选择你要的参数和你上下游引物的位置及你扩增产物的长度:出现Search Status窗口,点“OK”:出现Primer pairs窗口,#代表引物对的编号,依次为引物对所处的位置、产物的长度、最适合的退火温度、和GC的百分含量:点击任一行出现“PCR”窗口,告知你扩增片断的位置,最合适的退火温度等等信息:关掉“PCR窗口”和“primerPairs窗口”回到原来的窗口你就能看到你引物的序列和位置,图中手型鼠标所指即为引物序列:至此引物设计已经完成,你可以用“Analyse”菜单分析你的引物:有无引物二聚体、发卡结构等等:当上下游引物全选好以后,需要对引物进行评价并根据评价对引物进行修改。