定量校正因子的测定
- 格式:doc
- 大小:318.00 KB
- 文档页数:9

色谱的检测器对不同物质有不同的响应,换句话说,1mg化合物A在检测器上能产生1000mAu的响应,但同样是1mg的化合物B在该检测器上也许就只能产生847mAu的响应,所以我们不能在检测器输出1000mAu的响应时就认定样品中一定含有1mg化合物,这时就必须引入定量校正因子;校正因子的作用就是反映某物质的量与检测器响应之间的关系;定量校正因子分为两种:1.绝对定量校正因子f;f=M/A,其中M代表被测物质的量,A代表检测器信号响应,可以是峰面积或峰高,其意义为单位响应所反映的物质量;2.相对定量校正因子f';f'=fi/fs=Mi/Ai/Ms/As=MiAs/MsAi,其中i代表被测定物质,s代表选定的基准物质;绝对定量校正因子一般用于外标法,相对定量校正因子一般用于内标法;色谱法的含量测定中之所以要先用待测成分的对照品来建立校准曲线,然后才用这个曲线来计算待测样品中该化合物的含量,实际上就是在测定样品前先确定校正因子;日常操作中我们都是以:M标/A标=M样/A样直接计算样品含量了,所以没太注意有什么校正因子,事实上只要将公式作一个简单的变形:M样=A样M标/A标,不难看出式中的M标/A标其实正是定量校正因子f,那么M样=A样f了;简单来说可以理解为标准曲线的斜率最后提醒一点,用面积百分比法做含量测定时,不可简单地认为各成分的峰面积百分比就是它们的含量百分比哦,理由如上所述,各成分含量与响应的比例关系可不一定都相同啊色谱定量分析的依据是被测组分量与检测器的响应信号峰面积或峰高成正比;但是同一种物质在不同类型检测器上往往有不同的响应灵敏度;同样,不同物质在同一检测器上的响应灵敏度也往往不同,即相同量的不同物质产生不同值的峰面积或峰高;这样,各组分峰面积或峰高的相对百分数并不等于样品中各组分的百分含量;因此引入定量校正因子,校正后的峰面积或峰高可以定量地代表物质的量;1定量校正因子的定义定量校正因子分为绝对定量校正因子;由上述峰面积与物质量之间的关系W=fA可知:f=W/Af′称为绝对定量校正因子,即单位峰面积所代表的物质量;这是以峰面积表示的定量校正因子,也可以用峰高来表示定量校正因子;此外,也有用它们的倒数来表示的,简称为响应值;绝对定量校正因子的值随色谱实验条件而改变,因而很少使用;在实际工作中一般采用相对校正因子;其定义为某物质i与所选定的基准物质s的绝对定量校正因子之比,即:上式中w以重量表示,因此f又称为相对重量校正因子,通常称为校正因子f;本书均使用重量校正因子;如果物质量用摩尔表示,则称为相对摩尔校正因子,即f和f二者间的关系如下:2定量校正因子的测定气相色谱的定量校正因子常可以从手册和文献查到;但是有些物质的校正因子查不到,或者所用检测器类型或载气与文献的不同,这时就需要自己测定;测定时,准确称取待测校正因子的物质i纯晶和所选定的基准物质s,混合均匀后进样,测得两色谱峰面积A和A,用式求得物质i的相对重量校正因子;显然,选择不同的基准物质测得的校正因子数值不同;气相色谱手册中数据常以苯或正庚烷为基准物质;也可以根据需要选择其他基准物质,如采用归一化法定量时,选择样品中某一组分为基准物质;测定校正因子的条件检测器类型应与定量分析的条件相同;还应该注意的是,使用热导检测器时,以氢气或氦气作载气测得的校正因子相差不超过3%,可以通用,但以氮气作载气测得的校正因子与前二者相差很大,不能通用;而氢焰检测器的校正因子与载气性质无关;在一定操作条件下,分析组分i的质量m i或其在载气中的浓度是与检测器的响应信号色谱图上表现为峰面积A i或峰高h i成正比的,可写作:m i=fˊi·A i一、峰面积测量法1.峰高乘半峰宽法A=h·Y1/2 A=·Y1/22.峰高乘峰底宽度法3.峰高乘平均峰宽法A=h×4.峰高乘保留值法A= h·Y1/2=h·b·t R b可以约去,于是:A= h·Y1/2=h·t R5.积分仪二、定量校正因子色谱定量分析是基于被测物质的量与其峰面积的正比关系;但是由于同一检测器对不同的物质具有不同的响应值,所以两个相等量物质出的峰面积往往不相等,这样就不能用峰面积来直接计算物质的含量;为了使检测器产生的响应信号能真实地反映物质的含量,就要对响应值进行校正,因此引入“定量校正因子”quantitative calibration factor;m i=f’i A i 或f’I=m i/A i1.质量校正因子f m f m= =2.摩尔校正因子f M f M= = =f M·3.体积校正因子f V f V= = =f M4.相对响应值s’s’=1/f’三、几种常用的定量计算方法1.归一化法假设试样中有n个组分,每个组分的质量分别为m1,m2,…,m n, 各组分含量的总和m为100%,其中组分I的质量分数可按下式计算:w i = m i/m×100%= ×100%= ×100%若各组分的f值相近或相同,例如同系物中沸点接近的各组分,则上式可简式为:w i = ×100%该法优点是:简便、准确,当操作条件、如进样量、流速等变化时,对结果影响小; 2.内标法内标法是将一定量的纯物质作为内标物,加入到准确称取的试样中,根据被测物和内标物的质量及其在色谱图上相应的峰面积比,求出某组分的含量;例如要测定试样中组分i 质量为m i的质量分数w i ,可于试样中加入质量为m s的内标物,试样质量为m,则:n i=f i A i m s=f s A sm i/m s=A i f i/A s f s m i=A i f i/A s f s·m sw i=m i/m×100%= A i f i/A s f s·m s/m×100%一般常以内标物为基准,则f s=1,此时计算可简化为:w i=A i/A s·m s/m·f i×100%我配置了从甲烷、乙烷,乙烯,丙烷,丙烯,异丁烷,正丁烷,1-丁烯,顺-2-丁烯,反-2-丁烯,异丁烯,1,3-丁二烯的标准气;他们的体积分数也就是mol分数分别为:甲烷%乙烷%乙烯%丙烷%丙烯%异丁烷%正丁烷%1-丁烯%顺-2-丁烯%反-2-丁烯%异丁烯%1,3-丁二烯%,其余采用N2平衡%采用氧化铝毛细柱,FID检测器检测;采用单点矫正;那么计算各物质的校正因子只输入他们各自的mol含量就可调试安装师傅告诉我的,也就说甲烷的校正因子f甲烷=A甲烷峰面积;乙烷的校正因子f乙烷=A乙烷峰面积......,以此类推,得出各物质的校正因子;分析未知物时,假设未知全部检出,且不超过以上物种;那么各物质的含量为CH4%=f甲烷A'甲烷峰面积/ΣfiA'i,其余类似;不知道上面的说法是否正确,而且得出的是摩尔分数,对吗我从色谱书了解到,FID检测器为质量型检测器,那么峰面积可以代表哪些信息体积摩尔数质量摩尔体积分数质量分数是都可以,还是仅其中某个另外,校正因子产生后,当我检测未知物时,检测条件发生变化比如分流比,柱温,比如程序升温变化,进样器或者检测器,对校正因子是否有影响FID的峰面积乘以校正因子后表示质量,各物质的校正因子不同是因为检测器对各物质的响应强度不同;个人认为校正因子只与检测器的工作状态有关;采用相对校正因子法:计算的理论基础是带校正因子的面积归一化法1.配制含有水和乙醇的标准溶液,并记录以下参数,需要精密测定:标准溶液的体积,含有的水和乙醇的质量;简单一点配置就是用一个容量瓶,称重,加入少量水,再定量加入乙醇,加入的乙醇量按照体积百分比为80%的乙醇溶液估计;称重,然后用水定容至刻度,再次称重;2.精密量标准溶液和供试溶液进样3.分别计算水和乙醇相对校正因子;以乙醇校正因子为1.计算式如下:、水的相对校正因子=水的校正因子/乙醇的校正因子水的校正因子=标准溶液中水的重量/水的峰面积乙醇的校正因子=标准溶液中乙醇的重量/乙醇的峰面积乙醇的相对校正因子=水的校正因子/乙醇的校正因子=14.供试品水和乙醇的质量百分含量计算如下:水的质量百分含量=水的峰面积水的相对校正因子/水的峰面积水的相对校正因子+乙醇的峰面积乙醇的相对校正因子乙醇的质量百分含量=乙醇的峰面积乙醇的相对校正因子/水的峰面积水的相对校正因子+乙醇的峰面积乙醇的相对校正因子5.用上式计算,直接建立峰面积和质量百分含量的关系,相对校正因子只用定期测定一次即可;。

20111207栏目化药药物评价>>化药质量控制标题HPLC法校正因子研究中的几个问题作者张哲峰部门化药药学二部正文内容HPLC法具有将不同物质分离后逐一定量的分离分析能力,在药品有关物质检测中发挥着越来越重要的作用,成为药品杂质控制中常用而有效的手段之一。
在杂质对照品法、加校正因子的主成分自身对照法、不加校正因子的主成分自身对照法、峰面积归一化法等几种常用的杂质定量方式中,校正因子的研究对于选择合适定量方式,准确定量杂质具有重要意义,因而成为杂质分析方法研究中的重要内容之一。
但从目前注册申报资料实际情况来看,校正因子的研究和使用中尚存在一些需要进一步思考和关注的问题。
1.校正因子的定义及特点一般来讲,HPLC定量测定中,物质的检测量W与色谱响应值(峰面积等)A之间的比值称为绝对校正因子,即单位响应值(峰面积等)所对应的被测物质的量(浓度或质量);而某物质i与所选定的参照物质s的绝对校正因子之比,即为相对校正因子,即通常所讲的校正因子。
目前校正因子主要用于“加校正因子的主成分自身对照法”定量相关特定杂质,这种定量方式因考虑了杂质与主成分的绝对校正因子的不同所引起的测定误差,将标准物质的赋值信息转化为常数,固化在质量标准中,且不需长期提供标准物质,因而成为现阶段杂质控制较为理想可行的手段。
但这种方法有时会因不同仪器及色谱条件的波动,可产生一定范围的误差,需进行充分的方法耐用性验证,并结合色谱峰定位控制等措施,将误差控制在一定范围内。
2.校正因子的测定在校正因子的研究和使用中,标准物质、色谱条件、溶剂、检测波长等均是重要的影响因素,研究中需要予以关注。
2.1 校正因子的测定需要用到特定杂质及主成分的标准物质,这些标准物质应具备量值准确的特点,符合标准物质(对照品)的相关要求;其次,确定校正因子的分析方法应与最终确定的质量标准方法一致,色谱条件等需经筛选优化后确定,如有变更,需考虑对校正因子的影响,必要时重新确定;第三,要关注影响待测物UV吸收的各种因素,如溶液制备所用溶剂最好与最终确定的流动相相同,检测波长最好在特定杂质及主成分UV曲线的峰或谷处,避开吸收值急剧变化波段,以保证测定方法具有较好的耐用性,并保持测定结果的恒定。

Ⅰ、标准气体含量表示方法及换算一、标准气体含量表示方法1. 标准气体的摩尔分数(x b )(摩尔比)标准气体中组分气体B 的物质的量与标准气体中各组分物质的量的总和之比即为标准气体的摩尔分数。
此浓度是我们经常用的一种。
∑==ni iBB nn x 1式中:n B ─标准气体中组分气体B 的物质的量 n i ─标准气体中各组分物质的量的总和 常用10-2(%)、10-6表示2. 标准气体的质量分数(W B )(重量比)标准气体中组分气体B 的质量与标准气体中各组分的质量的总和之比即为标准气体的质量分数。
此也浓度是我们经常用的一种。
∑==ni iBB mm w 1式中:m B ─标准气体中组分气体B 的质量 m i ─标准气体中各组分质量的总和 常用10-2(%)、10-6表示3. 标准气体的质量摩尔浓度(mB )标准气体中组分气体B 的物质的量除以标准气体中各组分质量的总和为标准气体的质量摩尔分数。
ABB m n m =式中:n B ─标准气体中组分气体B 的物质的量 m A ─标准气体的总质量 常用mol/kg 、mol/g 、mmol/g 表示4. 标准气体的质量浓度(ρB ) 标准气体中组分气体B 的质量(m )除以标准气体的体积(v )和为标准气体的质量浓度。
Vm B =ρ式中:m ─标准气体中组分气体B 的质量 V ─标准气体的体积常用kg/m3、g/L 、mg/L ,ug/L 表示5.标准气体的物质的量浓度(c B )标准气体中组分气体B 的物质的量与标准气体的体积之比为标准气体的物质的量浓度。
Vn c BB =式中:n B ─标准气体中组分气体B 的物质的量 V ─标准气体的体积 常用mol/m3、mol/L 表示5. 标准气体的体积分数(ϕB )标准气体中组分气体B 的体积与标准气体中各组分物质体积的总和之比为标准气体的体积分数。
∑==ni iBB VV 1ϕ式中:V B ─标准气体中组分气体B 的体积 n i ─标准气体中各组分物质体积的总和常用10-2(%)、10-6表示,因为任何气体在标准状态下的摩尔体积均可近似为22.4L ,所以标准气体的体积分数可近似等于标准气体的摩尔分数。

实验B-23 定量校正因子的测定实 验 目 的1.掌握色谱定量校正因子的测定方法。
2.进一步熟悉,了解色谱仪的操作和性能。
实 验 原 理在一定色谱操作条件下,被测组分i 的重量(W i )与检测器的响应信号峰面积(A i ),成正比,W i =f i ′²A i ,这就是色谱定量分析的依据,式中f i ′为比例常数,称为被测组分i 的绝对校正因子。
由于检测器对不同物质具有不同响应,就不能用峰面积来直接计算物质的含量,而需要对响应值进行校正,这就是校正因子的意义,即f i ′= W i / A i ,可见f i ′代表了单位面积物质的重量。
由于f i 值与色谱条件有密切关系,不易准确测定,因而常采用相对校正因子f i ,即被测物质i 与标准物质s 的绝对校正因子之比(通常把“相对”二字略去):is si s s i s i i A W A W A W A W f f f i ∙∙===//'' 式中f s ′、 W s 、A s 分别为标准物质的绝对校正因子,重量及峰面积。
仪器与试剂1.仪 器岛津GC-14气相色谱仪 载气钢瓶H 2 色谱柱(参见前一实验) 微量注射器10μL2.试 剂101白色硅烷化担体(60-80目) 有机皂土34 邻苯二甲酸二壬酯 苯(分析纯) 甲苯(分析纯) 邻二甲苯(分析纯)实 验 步 骤1.实验条件:检测器:热导池检测器(TCD ) 桥电流100mA 温度:色谱柱 90°C 检测器 110°C 汽化室 150°C 载气: H 2纸速: 30 mm/min 进样量: 4μL2.色谱操作:(1) 准确称取苯0.4g(±0.0001g),甲苯0.4g(±0.0001g),邻二甲苯0.5g(±0.0001g)于具塞试管中,摇匀备用。
(2) 吸取混合试样4μL 进样,得到各组分的色谱图,出峰顺序为苯,甲苯,邻二甲苯。
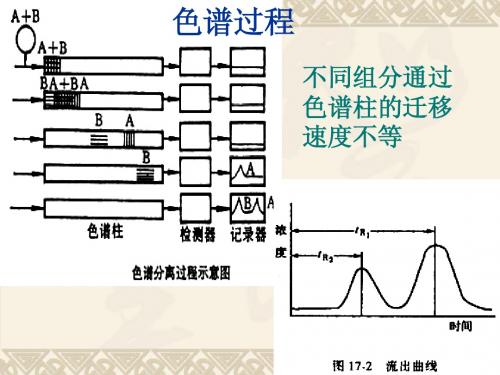

色谱定量分析色谱分析的重要作用之一是对样品定量。
色谱法定量的依据是:组分的重量或在载气中的浓度与检测器的响应信号成正比。
在此,响应信号指峰面积或峰高,表示为:i i i A f w =,其中:w i 为欲测组分i 的量,A i 为组分i 的峰面积,f i 为比例系数,在此称为校正因子。
由此可见,要准确定量,首先要准确测出峰面积与定量校正因子。
一、峰面积的测量1. 对称峰面积的测量对称色谱峰近似地看作一个等腰三角形,按照三角形求面积的方法,峰面积为i w h A h i i 2=,经验证明该方法计算的面积只有实际面积的0.94倍,故再乘一系数1.065,i w h A h i i 2065.1=,这是目前应用较广的计算法。
2. 不对称峰面积的测量在色谱分析中,经常会遇到不对称峰,多数不对称峰为拖尾峰,峰面积的计算方法为:取峰高0.15倍处和0.85倍处峰宽的平均值,乘峰高:h W W A h h ⨯+=)(2185.015.0 3. 大色谱峰尾部的小峰面积的测量分析某主成分中痕量组分时,常会遇到主峰未到基线,杂质峰开始馏出的情况。
此时,杂质峰面积计算法如下:沿主峰尾部划出杂质峰的基线,由峰顶作主峰基线的垂线。
峰顶为A ,垂线与主峰尾部交点为B ,峰高一半处峰宽为b ,则A=AB·b 。
4. 基线漂移时峰面积的测量基线漂移时的峰面积,形状与大峰后面拖尾的小峰的峰缝相似,计算方法相同。
5. 重合峰面积的测量在色谱分析中,常会遇到分离不完全的重合峰,峰面积可如下计算:两峰重合,如果交点位于小峰半高以下,可由峰高乘半高峰宽法计算两峰面积。
如果两峰交点位于小峰半高以上,通常是由交点作基线的垂线,再用剪纸称重法计算。
6. 峰高乘保留时间法同系物间,半高峰宽与保留时间呈线形关系:a bt W R h +=2,对于填充柱0≈a 。
当色谱峰很尖、很窄、半高峰宽不易测准时,可用保留时间代替半高峰宽R bt h A ⋅=065.1。
色谱定量分析中校正因子的使用在药物研发和QC岗位工作的人员在进行杂质定量时会经常遇到校正因子。
那么定量过程中为什么要使用校正因子、校正因子该怎么计算、得到的校正因子结果该怎么进行使用以及验证呢?下面小编将和大家一一进行分析这些问题,让大家透彻的了解校正因子。
1、为什么要使用校正因子?问题1:在做有关物质质量研究控制时,获得杂质是最让人头疼的一个问题,因有些杂质很难制备、稳定性差或者价格昂贵,难以长期提供杂质进行后续检测。
解决办法:因物质通过检测器时会有一个响应值,所以使用峰面积进行反应待测组分的含量就是一个很好的方法。
问题2:由于同一检测器对不同物质的响应值不同,所以当相同浓度的不同物质通过检测器时,产生的峰面积不一定相等,这种情况下使用峰面积进行反映待测组分的含量就会出现误差。
解决办法:为了消除这个误差,需要加入一个校正值,使得相同浓度的不同物质通过检测器时,产生的峰面积相等,以达到使用峰面积准确反映待测组分的含量,这个校正值就是我们常提到的校正因子。
举例如下:0.1mg/ml API的峰面积5000.1mg/ml 杂质峰面积是250测定某样品时检出API峰面积为500,待测组分为5。
当使用峰面积(面积归一化法)计算杂质的含量:5/500*100=1%当使用外标法进行计算杂质的含量:5*0.1/250/0.1*100=2%这样使用面积归一化法和外标法计算杂质结果就出现了误差。
当引入校正因子:500/250=2,进行计算杂质的含量:5*2/500*100=2%此时计算的结果就相吻合了。
以上就是我们在样品杂质定量时需要使用校正因子的原因。
2、校正因子的含义校正因子分为绝对校正因子和相对校正因子。
绝对校正因子:物质的检测量W与色谱响应值(峰面积等)A之间的比值相对校正因子:某物质i与所选定的参照物质s的绝对校正因子之比通常我们在实验过程中使用的就是相对校正因子,经常查阅USP药典的朋友会发现USP质量标准中使用的是响应因子,它是校正因子的倒数。
定量校正因子的测定
【色谱世界】【本书目录】【引用网址】/books/C/71/0.html
1. 绝对校正因子
由此方法测定出的校正因子称为绝对校正因子,它只适用于这一个检测器。
因为即使是换一个同一类型的检测器,甚至是换一个同一厂家生产的同一型号检测器,由于两个检测器的灵敏度总是有些差异的,这就使等量的同一种物质在这两个检测器上的响应值有所不同,因此计算出的绝对校正因子也有所不同。
同一个检测器,随着使用时间和操作条件改变灵敏度也在改变。
这些都使绝对校正因子在色谱定量分析中的使用有很大的局限性,为此引出了相对校正因子的概念。
2. 相对校正因子
常用的基准物质对不同检测器是不同的,热导检测器常用苯作基准物质,氢焰离子化检测器则常用正庚烷作基准物质。
通常人们将相对校正因子简称为校正因子,它是一个无因次量,数值与所用的计量单位有关。
根据物质量的表示方法不同,校正因子分为:
3. 峰高定量校正因子
在用峰高进行色谱定量时要使用峰高定量校正因子。
因为峰高定量校正因子受操作条件影响较大,因此一般不能直接引用文献值,必须在实际操作条件下,用标准纯物质测定。
对于同系物的峰高定量校正因子与峰面积定量校正因子间有如下的关系:
即可得到a,b值。
此方法不适于保留时间过小和不对称的色谱峰。
4. 响应值与校正因子的关系
响应值即为组分通过检测器时所产生的信号强度,可以用来表示检测器的灵敏度。
响应值与校正因子间有一定的关系。
即相对响应值为相对校正因子的倒数。
5. 校正因子的实验测量方法
准确称取色谱纯(或已知准确含量)的被测组分和基准物质,配制成已知准确浓度的样品,在已定的色谱实验条件下,取准确体积的样品进样,这样可以准确知道进入检测器的组分和基准物质的质量或摩尔数或体积,然后准确测量所得组分和基准物质的色谱峰峰面积,根据式(2-3-6)、式(2-3-7)和式(2-3-8),就可以计算出质量校正因子、摩尔校正因子和体积校正因子。
在没有合适的基准物质时,也可以测出绝对校正因子,利用绝对校正因子,在同一个检测器,相同的色谱实验条件下,也可作定量计算。
6. 确定校正因子的其他方法
除了上述利用实验方法直接测定校正因子之外,还可以利用已有的文献查找校正因子和利用一些规律估算校正因子。
(1)从文献上查找校正因子从文献上查找校正因子,主要是用于气相色谱的热导检测
器和氢火焰离子化检测器。
也有文献给出气相色谱的电子捕获检测器的响应值和校正因子,但电子捕获检测器与热导检测器和氢火焰离子化检测器不同,电子捕获检测器的响应值和校正因子与许多操作参数和检测器结构有关,如检测器结构尺寸,放射源种类,载气种类以及载气流速,检测器温度,极化电压大小,脉冲周期及脉冲宽度都对其响应值和校正因子有影响,它们之间存在相互依赖的复杂关系。
各个化合物特别是不同类型的化合物,在使用电子捕获检测器时都有各自的最佳操作条件。
因此,文献上提供的相对响应值和相对校正因子也受到操作条件和检测器性能的严格限制,一般说仅可作色谱定量校正的参考,最好通过实际实验进行测定。
对于液相色谱,检测器的校正因子在文献中是查不到的,这是因为液相色谱的分析条件变化较大,不易完全重复文献中的条件,故使用时要自己进行测定。
表2-3-6 和表2-3-7 给出了某些化合物在气相色谱的热导检测器和氢火焰离子化检测器上的响应值和校正因子。
表2-3-6 部分有机化合物在TCD上的校正因子(/books/C/403/0.html)
表2-3-7 部分有机化合物在FID上的校正因子(/books/C/404/0.html)
(2)利用规律对校正因子进行估算目前能对校正因子进行估算的只有气相色谱用的热导检测器和氢火焰离子化检测器。
当从文献中查不到适当数据,又没有已知准确含量的样品进行测定时,可按下述方法进行估算。
①热导检测器校正因子的估算:热导检测器的相对响应值一般只与组分、参比物质和载气性质有关,而与热导池结构、热丝温度、桥流、所用敏感元件以及柱温、载气流速等无关。
各类化合物在热导检测器上的响应值可按以下方法估算。
a. 同系物在热导检测器上的相对摩尔响应值(RMR)与其分子中的碳数或摩尔质量呈线性关系(图2-3-15),即
表2-3-8 同系物物质的G1与F1值(/books/C/406/0.html)
这一规律对于同系物中第一、二个成员偏差较大,但根据这一规律,如已知同系物中二个较高级成员的RMR值,就可推算其他成员的RMR值。
b. 基团截面积法,即某一化合物的RMR值,可由该化合物中各结构基团的RMR值加合而得,各种结构基团的RMR值见表2-3-9。
表2-3-9 各种基团的RMR值(以苯为100)(/books/C/407/0.html)
根据此规律估算乙酸丁酯的RMR值:
按此规律估算乙酸丁酯在热导检测器上的RMR值为134,试验值为135,说明估算结果与实验值能很好吻合。
此法多适用于极性化合物,如:酮、醇、醛、醚、酯和卤化物。
C. 同系物相对质量响应值与分子中碳原子数作图(图2-3-16),得一条渐近线似的关系曲线。
曲线在较高碳原子数时呈水平,趋向一个常数。
所以当组分中只有一些分子量相差不大,或只有较高分子量的组分时,可直接采用它们各自的面积百分数来计算百分浓度,即可认为各组分的校正因子相等。
当组分中既有低分子量组分,又有高分子量组分时,作定量计算就必须用相对质量校正因子加以校正(见本章第二节“定量方法”)。
d. 热导检测器的响应值与载气有关。
文献上给出的相对摩尔响应值(RMR)大多是以氦气(He)为载气,很少有氢气为载气的,我国国内色谱实验室多用氢气为载气,两种载气下的RMR值之间是否有关系呢?Mata等人在1963年提出了在以氦气为载气测得的RMR值换算成以氢气为载气时的RMR值的关系式:
但是,Rosie认为用氦气作载气时的RMR值与用氢气作载气时的RMR值差别不大,一般可通用,误差不超过3%,但用氮气(N2)作载气时相差就大了。
故一般用热导检测器时不用氮气作载气(用氮气作载气检测灵敏度也低很多),只用氢气或氦气作载气。
①氢火焰离子化检测器校正因子的估算:氢火焰离子化检测器的相对响应值与其结构——收集电极的形状和大小、喷口的粗细、极化电极的位置和形状有一定的关系。
主要是结构不同时,离子的收集效率和线性范围不尽一致,使得同一标准混合物,在结构不同的氢火焰离子化检测器上,测得的混合物中各组分的相对离子化效率有所不同。
氢火焰离子化检测器的相对响应值还与操作压力以及载气与燃气的流速有关,主要也是影响离子化效率。
所以,在给出相对响应值数据或使用文献值时,要注意氢火焰离子化检测器的结构、操作压力和载气与燃气的流速等实验参数。
若原始文献未能提供上述条件,而又要用其数据时,可用几个已知物,在自己的实验条件下,校对文献数据。
如能相符,则其他数据亦可应用;如不相符,可调节载气与燃气流速比等条件,尽量使自己的数据与文献值相符。
这样,其他数据在新确定的实验条件下也可使用。
虽然氢火焰离子化检测器的相对响应值受检测器结构、操作压力以及载气和燃气的流速影响,但与热导检测器一样,还是有一定的规律可遵循,也可进行估算。
a. 与热导检测器相同,同系物在氢火焰离子化检测器上的相对摩尔响应值(rmr)与其分子中的碳原子数呈线性关系(图2-3-17),可由式(2-3-14)表示:
表2-3-10 不同类型化合物的F,G,N o值(/books/C/408/0.html)
有效碳数。
表2-3-11 各种原子在不同类型化合物中所体现的有效碳数(/books/C/410/0.html)
例如,计算叔丁醇在氢火焰离子化检测器上的相对质量响应值,以苯为基准物质。
叔丁醇分子式为C3H9OH,有效碳数按表2-3-11所列各类化合物中各种原子的有效碳数计算:
实验值为0.68,两者相符。