一、共价键理论
- 格式:doc
- 大小:60.00 KB
- 文档页数:21

化学中的化学键理论化学键是指原子间的吸引力力,是分子形成的基础。
化学键的形成、性质和断裂是化学反应的重要环节,也是化学研究的核心内容。
化学键理论是化学学科中的重要分支之一,它揭示了化学键的性质和本质,为化学科学的发展和应用提供了理论基础。
1. 传统化学键理论在传统的化学中,原子间的化学键是指开尔文的“亲和力”理论。
它将原子的吸引力定义为原子核和共享了某些电荷的电子间的作用力,是一个纯经验的观点。
它不是一个特别准确的预测性理论,但是仍然在一些情况下被广泛使用。
2. 共价键理论共价键理论是指两个原子通过共享电子共同发展出的化学键。
这一理论揭示了共价键的本质,即原子间电子的共享。
共价键通常用杂化轨道理论来解释。
杂化轨道理论认为,原子的价电子空壳轨道中的电子可能会混合成新的、更稳定的轨道,称为杂化轨道。
杂化轨道提供了一个更准确的方法来描述共价键——如在氨分子中,氮原子价电子空壳轨道和氢原子的原子轨道混合,产生了四个杂化轨道,分别用于和四个氢原子组成共价键。
3. 离子键理论离子键理论是指形成离子键的原理。
它是一种典型的原子或分子排斥的现象。
当两种化学物质中含有带电离子时,离子间会产生电吸引力,因此导致它们结合到一起,而这些带电离子被称为离子。
离子键通常发生在化合物中,如氯化钠(NaCl)和硫酸二钾(K2SO4)。
4. 金属键理论金属键是指金属中的化学键,通常是由金属离子通过共享电子形成金属键。
金属离子在结晶中排列成空间有序的三维结构,形成晶格。
这种排列方式为金属提供了良好的机械性能和导电性能,在大规模制造工业用金属和合金方面有着重要的应用。
总之,化学键理论是化学学科的核心,它揭示了化学键的本质及其反应机理,为探索化学反应规律和推进实用化学技术发展提供了基础。
为了更好地掌握化学反应过程,我们需要深入了解化学键理论,并将其应用于实践中。

1927年,Heitler 和London 用量子力学处理氢气分子H2,解决了两个氢原子之间化学键的本质问题,并将对H2 的处理结果推广到其它分子中,形成了以量子力学为基础的共价键理论(V. B. 法)。
1共价键理论2共价键的形成A、B 两原子各有一个成单电子,当A、B 相互接近时,两电子以自旋相反的方式结成电子对,即两个电子所在的原子轨道能相互重叠,则体系能量降低,形成化学键,亦即一对电子则形成一个共价键。
形成的共价键越多,则体系能量越低,形成的分子越稳定。
因此,各原子中的未成对电子尽可能多地形成共价键。
配位键形成条件:一种原子中有孤对电子,而另一原子中有可与孤对电子所在轨道相互重叠的空轨道。
在配位化合物中,经常见到配位键。
、3共价键的特征——饱和性、方向性饱和性:几个未成对电子(包括原有的和激发而生成的),最多形成几个共价键。
例如:O 有两个单电子,H有一个单电子,所以结合成水分子,只能形成2个共价键;C最多能与H 形成4个共价键。
方向性:各原子轨道在空间分布是固定的,为了满足轨道的最大重叠,原子间成共价键时,当然要具有方向性。
1940年Sidgwick 提出价层电子对互斥理论,用以判断分子的几何构型. 分子ABn 中,A 为中心,B 为配体,B均与A有键联关系. 本节讨论的ABn 型分子中,A为主族元素的原子.4理论要点ABn 型分子的几何构型取决于中心 A 的价层中电子对的排斥作用. 分子的构型总是采取电子对排斥力平衡的形式.1)中心价层电子对总数和对数a)中心原子价层电子总数等于中心A 的价电子数(s + p)加上配体在成键过程中提供的电子数,如CCl4 4 + 1×4 = 8b)氧族元素的氧族做中心时:价电子数为6,如H2O,H2S;做配体时:提供电子数为0,如在CO2中.c)处理离子体系时,要加减离子价d)总数除以2 ,得电子对数:总数为奇数时,对数进1,例如:总数为9,对数为5 2)电子对和电子对空间构型的关系电子对相互排斥,在空间达到平衡取向. 3)分子的几何构型与电子对构型的关系若配体数和电子对数相一致,各电子对均为成键电对,则分子构型和电子对构型一致。






高中杂化轨道理论(图解)一、原子轨道角度分布图二、共价键理论和分子结构价键法(VB法)价键理论一:1、要点:⑴、共价键的形成条件:①、先决条件:原子具有未成对电子;②、配对电子参与成键的原子轨道要满足对称匹配、能量相近以及最大重叠的原则;③、两原子具有成单的自旋相反的电子配对,服从保里不相容原理。
⑵、共价键的本质:是由于原子相互接近时轨道重叠,原子间通过共用自旋相反的电子使能量降低而成键。
⑶、共价键的特征:①、饱和性,一个原子有几个未成对电子(包括激发后形成的未成对电子),便和几个自旋相反的电子配对成键;而未成对电子数是有限的,故形成化学键的数目是有限的。
②、根据原子轨道最大重叠原理,原子轨道沿其角度分布最大值方向重叠,即共价键具有一定的方向性。
⑷、共价键的类型:单键、双键和叁键。
①、σ键和π键。
ⅰ、σ键:沿键轴方向重叠,呈圆柱形对称,称为σ轨道,生成的键称为σ键σ是希腊字母,相当于英文的s,是对称Symmetry[`simitri]这个字的第一个字母)。
σ键形成的方式:ⅱ、π键:两个p 轨道彼此平行地重叠起来,轨道的对称面是通过键轴的平面,这个对称面就叫节面,这样的轨道称为π轨道,生成的键称为π键(π相当于英文的p ,是平行parallel[`p ?r ?lel]的第一个字母)。
π键的形成过程:,σ键和π键的比较 σ键(共价键中都存在σ键) π键 (只存在不饱和共价键中)重叠方式 (成建方向)沿两电子云(原子轨道)的键轴方向以“头碰头”的方式遵循原子轨道最大程度重叠原理进行重叠两互相平行的电子云(原子轨道)以“肩并肩”的方式遵循原子轨道最大程度重叠原理进行重叠 重叠程度重叠程度较大 重叠程度较小 电子云形状共价键电子云(重叠部分)呈轴对称 共价键电子云(重叠部分)呈镜像对称 牢固程度强度较大,键能大,较牢固,不易断裂 强度较小,键能较小,不很牢固,易断裂 化学活泼性不活泼,比π键稳定 活泼,易发生化学反应健 型项 目类型s-s、s-p、、p-p、s-SP杂化轨道、s-SP2杂化轨道、s-SP3杂化轨道、杂化轨道间p-pπ键,、p-p大π键是否能旋转可绕键轴旋转不可旋转,存在的规律共价单键是σ键,共价双键有一个σ键,有一个π键;共价叁键有一个σ键,有两个π键。

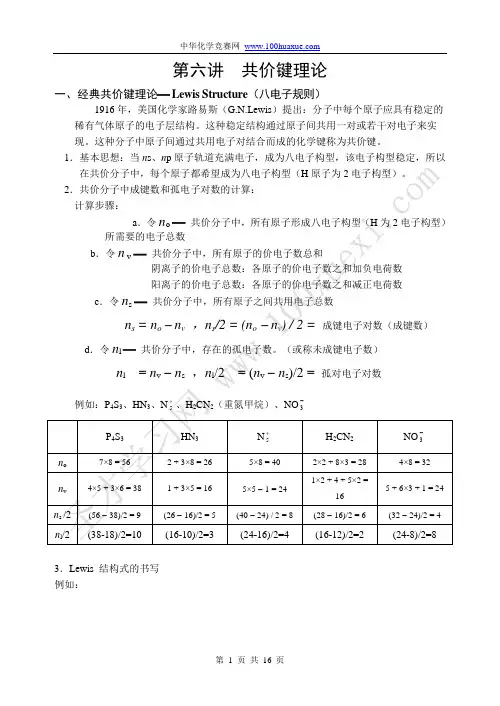
第六讲共价键理论一、经典共价键理论− Lewis Structure(八电子规则)1916年,美国化学家路易斯(G.N.Lewis)提出:分子中每个原子应具有稳定的稀有气体原子的电子层结构。
这种稳定结构通过原子间共用一对或若干对电子来实现。
这种分子中原子间通过共用电子对结合而成的化学键称为共价键。
1.基本思想:当n s、n p原子轨道充满电子,成为八电子构型,该电子构型稳定,所以在共价分子中,每个原子都希望成为八电子构型(H原子为2电子构型)。
2.共价分子中成键数和孤电子对数的计算:计算步骤:a.令n o−共价分子中,所有原子形成八电子构型(H为2电子构型)所需要的电子总数b.令n v−共价分子中,所有原子的价电子数总和阴离子的价电子总数:各原子的价电子数之和加负电荷数阳离子的价电子总数:各原子的价电子数之和减正电荷数c.令n s−共价分子中,所有原子之间共用电子总数n s = n o - n v,n s/2 = (n o- n v) / 2 = 成键电子对数(成键数)3.Lewis 结构式的书写例如:N 5+,,,NN N NN NN N NN NN N NN NN N N NCH 2N 2(重氮甲烷) ,HCHNN HC HNN(有时,孤对电子省略不写。
)练习:下列各LewisA.N O OOB. NO OOD.N O OO当Lewis 4.Lewis 结构式稳定性的判据 −− 形式电荷Q F (1) Q F 的由来: 以CO 为例nn v = 4 + 6 =10 n l / 2 = (10 - 6) / 2 = 2O 原子上的一个价电子转移给 ,所以氧原子的Q F 为+1,碳原子的Q F 为-1。
- 孤电子数2 = -1 Q F(O) = 6 -3 - 2 = +1H N N NH N N 02H N N N 0(I)(II)(III)形式(I)、(III)中形式电荷小,相对稳定,而形式(II)中形式电荷高,而且相邻两原子之间的形式电荷为同号,相对不稳定,应舍去。

化学键的价键理论共价键价电子对化学键是化学反应中最基本的概念之一,它描述了原子之间的结合方式。
化学键的形成涉及到共享或转移电子,其中共价键是最常见的一种化学键类型。
共价键形成的基本单位是价电子对,本文将探讨化学键的价键理论及共价键价电子对的性质。
1. 共价键的概念共价键是原子之间通过电子的共享形成的化学键。
在共享过程中,原子通过共享价电子对形成共价键,使得原子能量降低并达到更稳定的状态。
共价键可以形成在同种元素之间(如氢气分子H2)或不同元素之间(如氧气分子O2)。
2. 价键理论2.1 原子轨道和价电子对根据价键理论,原子由核心和围绕核心的电子组成。
电子存在于不同的轨道中,其中价电子是参与形成化学键的电子。
价电子对是共价键的基本单位,可以是一个或多个共享的电子对。
2.2 原子轨道的杂化原子的轨道通过杂化可以重新组合成新的轨道,以适应共享电子对的形成。
常见的杂化类型包括sp、sp2和sp3杂化。
sp杂化产生线性共价键,sp2杂化产生三角平面共价键,sp3杂化产生四面体共价键。
2.3 共价键的形成共价键的形成通过轨道重叠实现。
共价键的形成有两种基本的重叠方式:头-头重叠和边-边重叠。
在共价键形成中,电子云发生重叠,形成化学键。
3. 共价键价电子对的性质3.1 共价键的长度和键能共价键的长度取决于原子核间的距离,较短的键对应于较强的共价键。
共价键的键能是破坏化学键所需的能量,与键的强度相关。
3.2 共价键的极性共价键可以是非极性的或极性的。
非极性共价键是由相同或相似元素之间的共享电子对形成的,如氢气分子H2。
极性共价键是由不同元素之间的共享电子对形成的,电子云偏向电负性较高的原子。
3.3 共价键的结构和分子形状共价键决定了分子的结构和形状。
共价键的方向性以及原子杂化方式对分子形状产生重要影响。
杂化sp3形成四面体分子结构,sp2杂化形成平面三角形分子结构。
4. 应用和意义共价键的理论对于解释化学反应、分子形状及性质具有重要意义。
分子结构和共价键理论分子结构是指分子中原子之间的几何排列和相对位置。
分子结构的确定对于理解分子的性质和反应机制至关重要。
根据分子结构的不同,分子可以分为线性、平面三角形、正四面体、平面四边形、平面五边形、八面体等各种类型。
而共价键是指通过共用电子对来连接原子的一种化学键。
共价键的形成是原子间电子的重叠和共享,通过共享电子形成的共价键的强度与原子间的距离和电子云的重叠程度有关。
根据电子对的数量和形式,共价键又可分为单键、双键、三键等不同类型。
共价键理论是用来解释共价键形成和分子结构的理论体系。
共价键理论最初由路易斯在1916年提出,由于其简单和直观的描述方式,被广泛接受和应用。
根据共价键理论,原子通过共享电子对来完成对外层电子的填充,以达到稳定的电子结构。
共价键的形成遵循八个原则,即凯库勒原则,也被称为共价键的“八个原则”。
凯库勒原则的具体内容有:1.原子通过共享电子对来完成稳定的电子结构。
2.原子中的电子仅能拥有共价键所需的电子对数。
3.每个电子对对应一个共价键。
4.共价键通常与共价键的长度成正比,共价键越长,键能越小。
5.共价键的长度与原子半径和离子半径有关。
6.共价键的强度与键能成正比,共价键越紧密,键能越大。
7.共价键的强度与电子云的重叠程度有关,重叠程度越大,共价键越强。
8.共价键的强度与原子质量有关。
根据共价键理论,可以解释分子的稳定性、电荷分布、偶极矩和分子极性等性质。
分子的稳定性与共价键的强度和长度有关,共价键越紧密、越短,分子越稳定。
分子的电荷分布与原子间电子的共享程度有关,共价键的形成使得电子密度在分子中产生偏移,形成电荷云的分布。
分子的偶极矩和分子极性与分子中原子的电负性差有关,原子对电子的吸引能力差异越大,分子的偶极矩越大。
除了凯库勒原则,还有一些额外的因素可以影响共价键的形成和分子结构的稳定性,如共振、键角张力和立体位阻等。
共振是指分子中的双键或三键的位置可以在不同原子之间变化,形成多个共振结构,增加了分子的稳定性。
化学键的共价性与价键理论化学键是分子固有的结构特征之一,它决定了物质的性质以及化学反应的进行。
化学键的共价性与价键理论是化学中一个重要的概念,对于我们理解化学反应机制和性质变化具有重要意义。
共价性是指两个原子通过共享电子而形成的化学键的特性。
共价键形成的基本原理是原子通过共享其外层电子来使自己更加稳定。
在共价键中,原子通常会共享自己外层轨道上的电子,通过共享电子而形成共享电子对。
共价键的稳定性取决于电子的共享程度,即电子对的有效撞击面积越大,共价键越稳定。
电子对的有效撞击面积取决于两个原子的大小和形状以及电子密度,而这些又受到分子内外的其他电子的影响。
价键理论是解释共价键形成的一种理论。
它是根据分子轨道理论和原子轨道重叠概念来解释共价键的稳定性和性质的。
根据价键理论,当两个原子接近时,它们的原子轨道会重叠形成一个共价键。
这个过程中,原子轨道之间的电子会进行混合重叠,形成分子轨道。
分子轨道比原子轨道更稳定,所以共价键形成后分子比原子更加稳定。
根据原子轨道的对称性,可以分为σ键和π键。
σ键是最稳定的共价键,它是通过两个原子正面相互重叠形成的。
两个原子的轨道相互重叠后,形成了一个对称分子轨道,这个轨道的电子密度在原子间最大。
σ键的形成需要两个原子轴线上的电子重叠,通常形成于原子间成本占据空间最小的轨道上。
在化学反应中,σ键往往是最稳定的键,也是最不容易被断裂的键。
π键是一个相对不稳定的键,它是通过两个原子侧面相互重叠形成的。
两个原子的轨道相互重叠后,形成了一个非对称的分子轨道,这个轨道的电子密度主要在原子轴线的两侧。
由于π键的轨道在原子轴线的两侧,所以π键会受到旁边其他电子的排斥而不稳定。
在化学反应中,π键常常容易被破坏,因为它的结构相对较弱。
总而言之,共价性与价键理论是化学键的一个重要概念,它们描述了原子间通过共享电子形成键的过程和稳定性。
共价键的稳定性取决于电子的共享程度,而价键理论通过分子轨道理论和原子轨道重叠概念来解释了共价键的形成和性质。
什么是共价键理论
共价键理论是一种描述化学键的理论,主要内容如下:
1. 共价键的本质是电性的,共价键的结合力是两个原子核外共用电子对形成负电区域的吸引力,而不是正、负离子的之间的库伦作用力。
2. 如果A、B两个原子各有一个未成对的电子,若两个单电子所在轨道对称性一致则可以互相重叠,电子以自旋相反的方式成对,两原子形成共价单键,体系的能量降低。
3. 共用电子对也可由其中一个原子提供,称为共价配键。
4. 共价键具有方向性和饱和性。
方向性成因是原子轨道分布具有方向性,饱和性成因是原子价层的单电子数有限。
5. 共价键的特征可以用键能、键长、键角等几个物理量来描述,键角决定几何构型。
以上是共价键理论的主要内容,此理论主要解决H2分子成键实质的问题。
如需了解更多信息,建议查阅化学领域相关书籍文献或咨询化学领域专业人士。
一、共价键理论1.1、价键理论价键的形成是原子轨道的重叠或电子配对的结果,如果两个原子都有未成键电子,并且自旋方向相反,就能配对形成共价键。
例如:碳原子可与四个氢原子形成四个C—H键而生成甲烷。
HH..*.C4HHH*HHC+**.*HH由一对电子形成的共价键叫做单键,用一条短直线表示,如果两个原子各用两个或三个未成键电子构成的共价键,则构成的共价键为双键或三键CCCC双键三键共价键形成的基本要点:(1)成键电子自旋方向必需相反;(2)共价键的饱和性;(3)共价键的方向性——成键时,两个电子的原子的轨道发生重叠,而P电子的原子轨道具有一定的空间取向,只有当它从某一方向互相接近时才能使原子轨道得到最大的重叠,生成的分子的能量得到最大程度的降低,才能形成稳定的反之。
重叠最大H稳定结合Cl(1)H重叠较小 + 不稳定结合Cl (2)H(1s)Cl(2p)H不能结合(3)ClS 和P电子原子轨道的三种重叠情况1.2、分子轨道理论分子轨道理论是1932年提出了来的,它是从分子的整体出发去研究分子中每一个电子的运动壮态,认为形成的化学键的电子是在整个分子中运动的。
通过薛定谔方程的解,可以求出描述分子中的电子运动状态的波函数ψ,ψ称为分子轨道,每一个分子轨道ψ有一个相应的能量E, E近似的表示在这个轨道上的电子的电离能。
基本观点:(1)当任何数目的原子轨道重叠时,就可形成同样数目的分子轨道。
例如:两个原子轨道可以线性的组合成两个分子轨道,其中一个比原来的原子轨道的能量低,叫成键轨道(由符号相同的两个原子轨道的波函数相加而成),另一个是由符号不同的两个原子轨道的波函数相减而成,其能量比两个原子轨道的能量高,这中种分子轨道叫做反键轨道。
-=ψ(反键轨道)ψψ2ψA 2B能ψψ(原子轨道) AB量+=ψ1ψψ(成键轨道)1ψBA分子轨道能级图(2)和原子轨道一样,每一个分子轨道只能容纳两个自旋相反的电子,电子总是优先进入能量低的分子轨道,在依次进入能量较高的轨道。
(3)由原子轨道组成分子轨道时,必须符合三个条件:(a)对称匹配——既组成分子轨道的原子轨道的符号(位相)必须相同;(b)原子轨道的重叠具有方向性;(c)能量相近。
++ 成键轨道(Π)+--能量+-*反键轨道(Π ) +-+П轨道的示意图1.3、共价键的断裂有机化合物发生化学反应时,总是伴随着某些化学键的断裂和新的共价键的形成,共价键的断裂有两种断裂方式。
1、均裂——成键的一对电子平均分给两个原子或原子团,生成两个自由基。
A:B ? A• + B•(自由基)在有机反应中,按均裂进行的反应叫做自由基反应。
2、异裂——成键的一对电子在断裂时分给某一原子和原子团,生成正负离子。
(1) +CX(1)碳正离子C : X (2)(2) XC碳负离子在有机反应中, 按异裂进行的反应叫做离子型反应。
亲电反应由亲电试剂进攻而引发的反应。
离子型反应亲核反应由亲核试剂进攻而引发的反应。
亲电试剂——在反应过程中接受电子的试剂称为亲电试剂。
亲核试剂——在反应过程中能提供电子而进攻反应物中带部分正电荷的碳原子的试剂。
(二)、取代反应取代反应分自由基取代、亲电取代和亲核取代三种反应类型,其中烷烃的卤代反应是典型的自由基取代反应,苯环的卤代反应是亲电取代反应,而卤代烃的水解则是亲核取代反应的典范。
通常情况下,自由基反应要在引发剂存在的条件下才能进行,有时还需要光照或加热条件;芳烃的亲电取代需要催化剂;卤代烃水解是在碱性条件下进行的。
1.4、甲烷的自由基取代反应horυ?Cl Cl2 Cl ??链引发 CHCHHClCl34??CHCH-ClClCl3?3?2HCl-ClCHCH-ClCl2????3 ?链增长阶段CH-ClCH-Cl?ClCl22?22 ........................................................... ................................................................................ ..ClClCl??2CH-CHCHCH链终止阶段 333??3CHClCH-Cl?3?3从上可以看出,一旦有自由基生成,反应就能连续的进行下去,这样周而复始,反复不断的进行反应,故又称为链锁反应。
凡是自由基反应,都是经过链的引发、链的传递、链的终止三个阶段来完成的。
1.5、苯的亲电取代反应1.5.1、亲电取代反应的机理1、硝化反应+ O-NO] +SOH[HHONO + HOSOOH22422 O- H2NO 2H +- HNO2 NO+NONO222π络合物σ络合物+ HSOH +HSO4 242、卤代反应H-Br-BrBrHClFeBr[FeBr] 34Br2Br+ FeBr3π络合物σ络合物3、磺化反应+ SO + HO + HSO2 HSO33424OH +SOH3Hδδ OSOS+3O4、付—克烷基化(F-C)反应δδ +CHBrAlClCH+CH--Br......AlClCHCHCH[AlClBr]323 323323CHCH23+ CHCHH HBr23H+CHCH +32 AlCl31.5.2、苯环上定位规则的解释(1)、对间位基的解释 (以硝基苯为例)aO > N > C 由于电负性,因此硝基δ δOI为强吸电子基,具有效应,使苯环钝化。
δδ Nδb硝基的π键与苯环上的大π键形成π—π共轭,O δδ因硝基的强吸电子作用,使π电子向硝基转移,形成吸δC电子的共轭效应。
-I、-C方向都指向苯环外的硝基(电荷密度向硝基分布)使苯环钝化,因间位的电荷密度降低的相对少些,故新导入基进入间位。
硝基苯苯环上的相对电荷密度为:NO20.79δ0.95δδ 0.61(2)、对邻、对位基的解释a、甲基和烷基H3C sp为 23HHCCH >spsp3电负性2 具有σ—π共轭C sp为电荷移向苯环+ Cδ有效应 + I甲基具有δ δ诱导效应+I和共轭效应+C都使苯环上电子云密度增加,邻位增加的更多,量子化学计算的结果如下:CH0.9631111.0171 10.99911.011总值为6总值大于6故甲基使苯环活化,亲电取代反应比苯易进行,主要发生在邻、对位上。
b、具有孤电子对的取代基(-OH、-NH、-OR等) 2以苯甲醚为例:OCH 3O > CI电负性具有效应δ δ+ CP —π氧上的电子对与苯形成共轭,具有效应δ由于+C > -I,所以苯环上的电荷密度增大,且邻、对位增加的更多些,故为邻对位定位基。
1.6、卤代烃的亲核取代反应1.6.1、单分子亲核取代反应(S1反应) N实验证明:3?RX CH=CHCHX 苄卤的水解是按S1历程进行的。
22NCHCH33 --CBr+ OH+ BrCOHCH CH33CHCH33 V = KCH C - Br( 3 3)因其水解反应速度仅与反应物卤代烷的浓度有关,而与亲核试剂的浓度无关,所以称为单分子亲核取代反应(S1反应)。
N1、反应机理S1反应是分两步完成的。
第一步: NCH2CHCH33 -δδ慢+ BrCBrCBrCCHCH……CH 333CHCHCH3331( )过渡态第二步:CH2CHCH33 δ快-δCCOHOHC+ OHCHCH……CH 333CHCHCH3332过渡态( )反应的第一步是卤代烃电离生成活性中间体碳正离子,碳正离子再与碱进行第二步反应生成产物。
故S1反应中有活性中间体——碳正离子生成。
N2、S1反应的立体化学 N(1)、外消旋化(构型翻转 + 构型保持)2S1反应第一步生成的碳正离子为平面构型(正电荷的碳原子为sp杂化的)。
N 第二步亲核试剂向平面任何一面进攻的几率相等。
R1RRR11 1CBrCC+OHHO CabRRRRR2232 2RRR333HOa构型转化b 构型保持外消旋体CNu如:HHHHCl OH+CCC+CClCHOHCHHOOCHH慢 3323CHCHCHCHCH 653656565(2)、部分外消旋化(构型翻转 > 构型保持)S1反应在有些情况下,往往不能完全外消旋化,而是其构型翻转 > 构型N 保持,因而其反应产物具有旋光性。
n-n-CH CHn-613CH613613HO60%乙醇2 HOCBrCOHC+S1H HN条件HCHCH33 CH3( ) - 2 -( ) - 2 - ( ) - 2 -辛醇溴辛烷辛醇67%33%左旋2-溴辛烷在S1条件下水解,得到67%构型翻转的右旋2-辛醇,33%构型N 保持的左旋2-辛醇,其中有33%构型翻转的右旋2-辛醇与左旋2-辛醇组成外消旋体,还剩下34%的右旋2-辛醇,所以,其水解产物有旋光性。
1.6.2、双分子亲核取代反应(S2反应) N实验证明:伯卤代烷的水解反应为S2历程。
N- -RCHOH + BrRCHBr + OH22- V = K[ RCHBr ] [ OH ]2 V =水解速度K =水解常数-因为RCHBr的水解速率与RCHBr和OH的浓度有关,所以叫做双分子亲22 核取代反应(S2反应)。
N1、反应机理新键的形成和旧键的断裂同步进行,无中间体生成,经过一个不稳定的―过渡态‖。
HHH δδ HO C BrHO +…+ BrCCBrHO…HH HHHH过渡态中心碳原子与五个从离去基团溴原子的HO其他原子或基团相连接,背面进攻中心碳原子,受溴原由于较为拥挤,导致其热力学稳定性差,易于子的电子效应和空间效应的影断键,使中心碳原子恢响最小。
3sp复杂化。
其反应过程中的轨道重叠变化如下图所示:HHHHO+ BrBrCHOCBrHOC HHHHHHS2 反应成键过程中轨道转变示意图N2、S2反应的立体化学 N-(1)、异面进攻反应(Nu从离去基团L的背面进攻反应中心)。
δδ-- Nu C LNuCNu C + L ……(2)、构型翻转(产物的构型与底物的构型相反——瓦尔登Walden转化)。
HC例如: 613HC613S2N Br+HOCHOC+ BrHH HCCH33 ( ) - 2 -( ) - 2 -溴辛烷辛醇= 34.2α= 9.9αCHCHCH613 613613δδ128 128CIIC++128ICIIH HICHCH33 CHH3实例说明,通过水解反应,手性中心碳原子的构型发生了翻转。
根据大量立体化学和动力学研究材料,可以得出下面的结论:按双分子历程进行亲核取代反应,总是伴随着构型的翻转。
也就是说,完全的构型转化往往可作为双分子亲核取代反应的标志。
(三)、加成反应加成反应的类型分亲电加成反应和亲核加成反应两种。