手性色谱分析..
- 格式:doc
- 大小:433.00 KB
- 文档页数:30


手性高效液相色谱法手性药物对映体在人体内与受体、酶等生物大分子互相作用,展现出复杂的对映体挑选性,进一步表现为不同的药理作用、代谢过程和毒性反应。
对于手性药物,可能其中一个对映体活性高、疗效好,为活性对映体(优对映体);另外的活性低甚至没有活性的为劣对映体。
因为劣对映体没有药效或药效较低,甚至可能产生严峻的不良反应,基于此,FDA 于1992年领先发布了手性药物指导原则,我国亦于2006年颁布了《手性药物质量控制讨论指导原则》。
为了评价手性药物的生物活性,监测对映体的光学纯度,建立和进展迅速精确的药物对映体分别办法具有重要的意义。
手性高效液相色谱法通常分为挺直法和间接法。
间接法又称手性衍生化试剂法(chiral derivatization reagent, CDR),是将对映体与手性光学试剂反应,生成一对非对映异构体之后以常规固定相分别,因此是对手性衍生物的非手性分别。
而挺直法则是采纳手性固定相法(chiral stationary phase, CSP)或将手性化合物加入到流淌相中,再用常规固定相分别的手性流淌相法(chiral mobile phase, CMP)。
手性固定相法因其用法简便快捷、重复性好、精确度高、应用范围广而倍受青睐。
一、手性色谱柱的分类目前,已商品化的手性固定相有100多种,按照手性固定相和溶剂的互相作用机制,Irving Wainer首次提出了手性色谱柱的分类体系:第1类:通过氢键、π-π作用、偶极-偶极作用形成复合物;第2类:既有1类中的互相作用,又存在包埋复合物。
此类手性色谱柱中典型的是由纤维素及其衍生物制成的手性色谱柱;第3类:基于溶剂进入手性空穴形成包埋复合物。
这类手性色谱柱中最典型的是环糊精型手性柱,另外冠醚型手性柱和螺旋型聚合物,如聚(苯基甲基甲基丙烯酸酯)形成的手性色谱柱也属于此类;第4类:基于形成非对映体的金属络合物,是由Davankv 开发的手性分别技术,也称为.手性配位交换色谱(CLEC);第5类:蛋白质型手性色谱柱,手性分别是基于疏水互相作用和极性互相作用得以实现的。
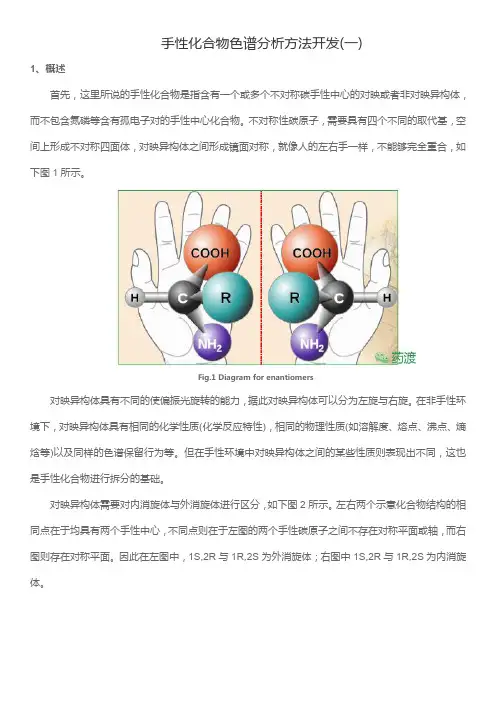
手性化合物色谱分析方法开发(一)1、概述首先,这里所说的手性化合物是指含有一个或多个不对称碳手性中心的对映或者非对映异构体,而不包含氮磷等含有孤电子对的手性中心化合物。
不对称性碳原子,需要具有四个不同的取代基,空间上形成不对称四面体,对映异构体之间形成镜面对称,就像人的左右手一样,不能够完全重合,如下图1所示。
Fig.1Diagram for enantiomers对映异构体具有不同的使偏振光旋转的能力,据此对映异构体可以分为左旋与右旋。
在非手性环境下,对映异构体具有相同的化学性质(化学反应特性),相同的物理性质(如溶解度、熔点、沸点、熵焓等)以及同样的色谱保留行为等。
但在手性环境中对映异构体之间的某些性质则表现出不同,这也是手性化合物进行拆分的基础。
对映异构体需要对内消旋体与外消旋体进行区分,如下图2所示。
左右两个示意化合物结构的相同点在于均具有两个手性中心,不同点则在于左图的两个手性碳原子之间不存在对称平面或轴,而右图则存在对称平面。
因此在左图中,1S,2R与1R,2S为外消旋体;右图中1S,2R与1R,2S为内消旋体。
Fig.2Name and distinguish between mesomer and racemate对于手性化合物的拆分,规模比较大的时候,可使用其他手性试剂(如酒石酸钠)与待拆分的化合物形成非对映异构体,然后根据非对映异构体之间具有不同的物理化学性质,进行相应的分离单元操作。
而在分析实验室中,一般是采用色谱法进行拆分,其中包括使用手性固定相法以及在流动相中添加手性流动相形成手性拆分环境的方式。
其中手性固定相拆分法包括气相色谱以及液相色谱。
对于气相色谱拆分手性化合物,其拆分选择性主要取决于所使用的手性固定相的种类以及色谱分离的温度。
一般气相用于低沸点的手性化合物的拆分,对于有机酸碱等极性手性化合物的拆分,一般需要先进行柱前衍生化处理,使之形成相应的酯或者酰胺。
用于气相手性拆分的手性固定相均为环糊精衍生物类,包括β以及γ环糊精,α环糊精比较少;其最高耐受温度不会超过220℃,而且分离温度超过120℃的时候,固定相的手性选择性开始降低;超过200℃的时候,固定相的手性选择性几近与无。

手性药物的分离在色谱法中的应用手性药物是指具有手性结构的药物。
它们可以分为左旋和右旋两种类型,两者化学性质相同,但左右旋异构体对生物系统的影响却截然不同,这种现象被称为手性诱导失活效应。
因此,在制药过程中需要对手性药物进行分离,以确保药效和安全性。
色谱法是分离手性化合物的主要方法之一,其基本原理是利用不同化合物的物理、化学性质差异,通过分离柱将混合物中的目标物分离出来。
以下是一些色谱法在手性药物分离中的应用。
手性高效液相色谱法(HPLC)手性HPLC是目前最常用于手性药物分离的方法之一,它是利用手性固定相在悬浊液中对手性化合物进行分离。
具有手性结构的固定相与目标分子相互作用,从而实现分离。
手性HPLC可以分别采用手性固定相或手性混合物来进行分离。
此外,在手性HPLC中,主要可以采用簇列技术或化学反应转化手性方法来提高分离效率和选择性。
毛细管电泳(CE)毛细管电泳是一种基于电化学原理的分离技术,它利用电场将样品中的分子分离。
在毛细管电泳中,可以采用手性高分辨涂层来进行手性药物的分离。
在此基础上,还可以采用手性化合物作为毛细管填充剂,进一步提高分离效率和分离度。
气相色谱法(GC)气相色谱法是一种利用气体作为流动相的色谱法。
在处理手性药物时,通常需要使用手性柱和手性混合物。
与HPLC不同,该方法的分离依赖于分子间的“挤压”力。
因此,手性柱具有不同的式样,以保证灵敏度和选择性。
超临界流体色谱法(SFC)SFC是一种介于HPLC和GC之间的色谱法。
它使用超临界流体作为移动相,可以在温度和压力条件下实现高效率的手性药物分离。
通常使用手性柱和手性对映异构体混合物进行分离。
此外,还可以应用具有特定分子功能的催化剂来提高分离效率。
总之,手性药物分离是一项非常复杂的任务,需要使用不同的色谱技术和方法来实现。
无论是HPLC、CE、GC还是SFC,它们都有各自的优缺点和适用范围,因此在选择分离方法时需要综合考虑样品特性,实验设备和分离效率与成本等因素。


有机化学的手性分析方法
在有机化学领域中,手性分析是一项十分重要的工作。
手性化合物是指分子的结构镜像不能完全重合的分子。
因此,手性分析的目的就是确定有机化合物中手性中心的配置。
在本文中,将介绍几种常用的手性分析方法。
一、圆二色谱分析法
圆二色谱分析法是一种利用圆二色现象测定有机物的手性的方法。
圆二色现象是指左旋光和右旋光通过具有手性的物质后,光传播方向不变,但相位差发生变化的现象。
通过观察物质在不同波长下的圆二色光谱,可以确定其手性。
二、红外吸收光谱分析法
红外吸收光谱分析法是一种常用的手性分析方法。
在红外光谱中,手性物质通常表现出特定的旋光效应,通过比较旋光贡献可以判断有机物的手性。
三、核磁共振分析法
核磁共振分析法是一种非常重要的手性分析方法。
通过核磁共振技术,可以观察到手性物质中的不对称中心周围原子核的信号差异,从而确定有机物的手性。
四、质谱分析法
质谱分析法是一种高灵敏度的手性分析方法。
通过质谱仪对有机物进行分析,可以观察到手性分子离子的不同质量谱峰,从而确定有机物的手性。
五、氨基酸序列分析法
氨基酸序列分析法主要用于蛋白质的手性分析。
通过氨基酸序列分析仪,可以确定蛋白质中的手性氨基酸的排列顺序,从而确定蛋白质的整体手性。
综上所述,有机化学的手性分析方法主要包括圆二色谱分析法、红外吸收光谱分析法、核磁共振分析法、质谱分析法以及氨基酸序列分析法。
这些方法各自有其优点和适用范围,科学家们可以根据具体情况选择合适的手性分析方法来进行研究。

色谱分析中的手性分离技术色谱分析是一种常见的分离和检测技术,它可以通过不同成分在色谱柱上的运移速度差异,实现样品中组分的分离。
而手性分离技术则是其中一种具有广泛应用的技术。
手性分离技术又称拆分体分离技术,是指将具有手性的化合物分离成其对映异构体的过程。
手性分离技术主要有两种:手性凝胶色谱和手性高效液相色谱。
手性凝胶色谱是一种传统的手性分离技术,它利用具有手性结构的聚合物凝胶作为色谱填料,通过样品分子与凝胶之间的分子识别作用实现分离。
手性凝胶色谱是一种相对简单的手性分离技术,但是由于其分离程度较低,通常用于对手性分析的初步筛查。
手性高效液相色谱是一种高效手性分离技术,它基于手性色谱填料的表面手性区分作用和反相分离作用,实现对手性化合物的高效分离。
在手性高效液相色谱中,手性色谱柱成为关键的分离工具,色谱柱内填充了各种具有手性结构的填料,如纳米结构材料、束缚配体、离子交换树脂等。
手性高效液相色谱技术需要精密的操作和控制技术,同时对手性填料的选择和性能也十分关键。
常见的手性高效液相色谱模式包括正相模式、反相模式和杂相模式。
正相模式下,填料是手性站点,流动相是水/有机溶剂混合物,溶液的极性越强,分离能力越高;反相模式下,填料是非手性的,分离基于无手性分子和手性分子与填料的相互作用,流动相是弱极性有机溶剂/水混合物;杂相模式是正相和反相模式的结合。
手性高效液相色谱技术在制药、化妆品、食品、医疗诊断等领域得到了广泛应用。
例如,在药物研发中,手性高效液相色谱可以对药物的对映异构体进行分离和鉴定,以确定对映异构体的药效和安全性;在食品领域,手性高效液相色谱可以对添加的手性能呈现不同风味的香料成分的组成比例进行分离和鉴定。
当然,手性分离技术也存在一些困难和局限性。
一方面,手性化合物的对映异构体之间的物理和化学性质非常相似,因此分离困难。
另一方面,手性化合物的分离需要精密的手性填料和色谱柱控制技术,手性柱的制备和使用成本也较高。

药物研究中手性分离分析方法及技巧手性药物是指药物分子结构中引入手性中心后,得到的一对互为实物与镜像的对映异构体。
液相色谱法成为目前手性药物分离测定的首选方法,根据实际工作中需要的手性分离问题,总结如下:1、流动相手性分析很关键的一项是流动相的选择,手性分析一般都采用正相,使用最多的流动相是正己烷、正庚烷、乙醇和异丙醇这四种,其中起洗脱作用的流动相是乙醇和异丙醇,正己烷和正庚烷用来调节流动相的洗脱强度。
正己烷和正庚烷对于样品分离没有什么太大的影响,不会改变选择性和分离度,通常都可以混用,不过正庚烷比正己烷对人体的伤害要小很多,但价格是后者的一倍,所以欧美的很多大制药公司多使用正庚烷,而国内多使用正己烷。
乙醇和异丙醇对样品的分离起关键的作用,不同的醇有不同的选择性,改变醇的种类可以改变选择性,常用的醇类是乙醇和异丙醇,甲醇不能使用是因为它和正己烷、正庚烷不互溶,叔丁醇粘度太大,一般作为添加剂配合乙醇或者异丙醇少量使用,提供特殊的选择性,通常能起到意想不到的效果。
一般情况下分析手性样品,很多人推荐首选异丙醇,但是我喜欢首选乙醇,因为乙醇气味比异丙醇好一点,且乙醇做流动相压力要低一些,实际上二者差别不是太大。
流动相里经常需要添加酸或者是碱来调节峰形,常用的酸有三氟乙酸、乙酸和甲基磺酸,碱一般是二乙胺和三乙胺,也有用乙醇胺和异丁胺的,流动相里添加酸和碱的浓度一般要求控制在0.2%(体积比)以下,我们一般用0.1%,使用的原则一般是酸性样品加酸,碱性样品加碱,但实际上很多样品是即含酸性基团又含碱性基团,这就要看哪个基团作用强了,对于某些含氨基的两性样品,例如苯甘氨酸,甲基磺酸是一个非常好的选择,磺酸基能够抑制氨基的碱性,又能提供一个酸性的流动相环境,使样品既能得到很好的分离又能获得对称的峰形。
一般做纯度分析检测杂质含量时我们要求尽量的采用低波长来让尽可能多的杂质有紫外吸收,而做手性分析时我们需要采用尽可能高的波长来去除在低波长下才有吸收的杂质的干扰,一般原则还是尽量选择样品紫外吸收最好的地方来获得较高的灵敏度,但流动相里添加二乙胺会导致在低波长下基线波动变大,系统难以平衡,这种情况下一般要提高检测波长,实际操作过程中有些样品在高波长下吸收非常差,只能用低波长检测,这样的样品可以尝试在样品稀释的时候加入过量的二乙胺(但不宜太多),而流动相用中性,从而获得满意的分析结果。

药物分析中的手性分析技术应用手性分析是药物分析领域中的重要技术之一。
由于药物分子中存在手性中心,即分子中存在手性异构体,其对于药物活性、代谢和药效等方面具有重要的影响。
因此,手性分析技术在药物研发、质量控制和临床应用中扮演着重要的角色。
本文将就药物分析中的手性分析技术应用进行论述。
一、手性分析技术概述手性分析技术是对手性药物的立体特性进行定性和定量分析的一类分析方法。
常见的手性分析技术包括极性手性色谱法(CSP)、核磁共振技术(NMR)和圆二色光谱技术(CD)等。
这些技术可以对手性药物进行手性异构体的分离和结构鉴定,进而研究手性药物的性质和应用。
二、极性手性色谱法(CSP)在药物分析中的应用极性手性色谱法是一种高效的手性分析方法,广泛应用于药物分析领域。
该方法利用手性色谱柱对手性异构体进行分离,通过优化色谱条件实现手性化合物的定性和定量分析。
极性手性色谱法在药物质量控制、药代动力学研究和药效学等方面发挥着重要的作用。
三、核磁共振技术(NMR)在手性分析中的应用核磁共振技术是一种基于核磁共振现象的手性分析方法。
通过测定手性异构体的化学位移差异,可以实现对手性异构体的身份鉴定和含量分析。
核磁共振技术具有无损、高灵敏度和高分辨率等优点,在药物分析中得到广泛应用。
四、圆二色光谱技术(CD)在手性分析中的应用圆二色光谱技术是对手性分子的光学旋光性质进行分析的一种有效手段。
通过测量手性化合物在紫外-可见光区域的旋光角度,可以确定手性异构体的构型和含量。
圆二色光谱技术具有高选择性和灵敏度,广泛应用于药物分子的手性分析和结构研究。
五、手性分析技术在药物研发中的应用手性分析技术在药物研发中起到了至关重要的作用。
在新药研发过程中,药物化学师需要对合成的手性药物进行手性分析,确定主要手性异构体的存在与含量,并进一步评估其药代动力学和药效学特性。
手性分析技术的应用使得药物研发人员能够更全面地了解手性药物的特性,指导药物设计和优化。
药物分析中的手性分析技术研究手性分析技术在药物分析中的研究药物是人类对抗疾病的重要工具,但很多药物都存在手性的特性。
手性分析技术的发展对于药物的研究与合成具有重要的意义。
本文将介绍药物分析中的手性分析技术及其研究进展。
一、手性与药物手性是化学中常见的现象,指的是分子存在两个非重叠的立体异构体,分别被称为左旋体和右旋体。
由于手性分子的空间结构不对称,其在生物体内的代谢与作用机制往往存在差异。
一种手性药物的两个异构体在生物作用上可能具有完全相反的效果。
因此,对手性药物的手性分析具有重要的理论和实践意义。
二、手性分析技术的原理在药物分析中,常用的手性分析技术主要包括气相色谱法(GC)、液相色谱法(HPLC)、毛细管电泳法(CE)等。
这些技术利用手性分离柱或手性分离剂作为分离介质,通过衡量手性分子的分离度来确定样品中手性异构体的相对含量。
1. 气相色谱法(GC)气相色谱法是一种常用的手性分析技术。
该技术利用手性柱通过手性相互作用实现手性分离。
常见的手性柱包括化学手性柱和拓展手性柱。
气相色谱法具有分离度高、分析速度快、准确性高的优点,广泛应用于药物分析中。
2. 液相色谱法(HPLC)液相色谱法是另一种常用的手性分析技术。
该技术主要利用手性分离剂与手性分析物之间的相互作用实现手性分离。
液相色谱法分离度较高,适用性广泛,常用于药物的手性分析及手性异构体的定量分析。
3. 毛细管电泳法(CE)毛细管电泳法是利用毛细管中的电渗流和电泳作用实现手性分离的一种分析技术。
该技术具有分离度高、样品消耗少等特点,适用于药物样品中手性异构体的分析与检测。
三、手性分析技术的应用手性分析技术在药物研究与开发中具有广泛的应用。
通过手性分析,可以评估药物的手性纯度、分离手性异构体、研究手性异构体的代谢过程等。
1. 评估药物的手性纯度药物合成过程中,常常会产生手性异构体的混合物。
通过手性分析技术,可以确定药物样品中各个手性异构体的相对含量,评估药物的手性纯度,确保药物的质量和疗效。
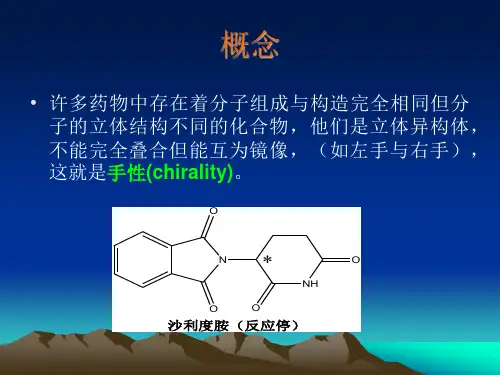
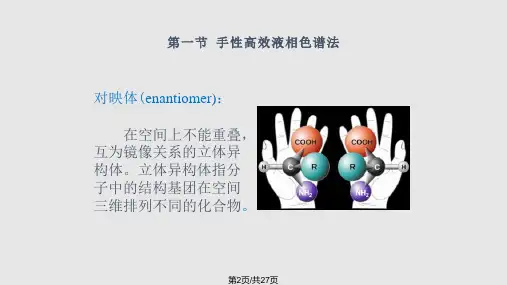
1手手性性高高效效液液相相色色谱谱法法**手手性性药药物物分分析析的的概概念念 **常常用用手手性性高高效效液液相相色色谱谱法法 手手性性衍衍生生化化试试剂剂法法 手手性性固固定定相相法法 手手性性流流动动相相添添加加法法2手手性性的的概概念念::一一种种镜镜像像反反射射的的对对称称性性3手性分子:组成相同但空间结构上互成镜像的分子,称之为对映异构体。
分子结构中含有不对称碳原子是最常见的手性结构。
根据对偏振光的作用不同可分为R、S体,两者的等量混合物称之为消旋体。
OH COOHHCH3OHCOOHHCH34Mirror Mirror手手性性异异构构体体在在药药理理学学效效应应上上的的差差异异 ● Pfeiffer 规则:● 对映异构体之间的生物活性存在着差异; ● 不同的对映体之间活性的差异是不同的;当手性药物的有效剂量越低,即药效强度越高时,则对映体之间的药理作用的差别越大。
外消旋体和其两种单一对映体是不同的3种实体! 5对对映映体体与与生生物物大大分分子子的的三三点点作作用用c abdabd cαγβαβγ手性分子的a 、b 、c 三个基团与受体分子的活性作用点、、结合,是高活性对映体(优映体)。
手性分子的a 、b 、c 三个基团中只有a 和b 与受体分子的活性作用点和结合,是低活性对映体(劣映体)。
6在未研究清楚两种单一对映体之间的生物学差异时,以消旋体给药往往会影响药物质量,甚至会严重损害人体健康。
“反应停”(Thalidomide)作为人工合成药,当时投入使用时是两种对映体的混合物。
7反应停:五十年恩怨发展趋势:劣映体本身或其代谢物产生毒副作用,不再使用外消旋体。
外消旋体转换成单一对映体,不仅提高质量,还延长药物寿命。
如:氧氟沙星的左旋异构体活性更强,左旋氧氟沙星临床使用剂量是消旋体的一半。
10手性拆分(Chiral resolution)●对映体除了偏振光的偏转方向不同外,其它理化性质完全相同,因而分离难度大。
●手性色谱拆分方法:创造(或引入)手性环境,构造非对映异构体,使药物对映体间呈现理化特性的差异,从而实现药物对映体的色谱分离。
.11组成相同但空间结构上互成镜像的分子,称之为对映异构体;不成镜像的分子,称之为非对映异构体,分子中所含手性碳越多,非对映异构体的数目越多.非对映异构体对映体对映体(- )盐酸伪麻黄(-)麻黄碱(+)麻黄碱(+ )伪麻黄碱(- )伪麻黄碱13 HOHCH3HCH3.H Cl色谱条件:色谱柱:苯基柱150mm×4.6mm,I.D. 5mm;流动相:三乙胺-水(含50mmol·L-1磷酸二氢钾,磷酸调pH至4.0)-甲醇(0.01∶90∶10, v/v/v);流速1.0 mL·min-1;检测波长:210nm;柱温:室温。
14手性高效液相色谱法(Chiral HPLC)●手性衍生化试剂法(CDR)(分析物)●手性固定相法(CSP)(固定相)●手性流动相添加法(CMP)(流动相)● 15手性试剂衍生化法(CDR ) ● 原理:光学活性药物与有高光学纯度的手性试剂于分离前衍生化,在药物对映体中引入另外一个手性,形成非对映异构体,再以HPLC 分析。
(R)SA (R)SE (R)SA(R)SE (S)SA (R)SE (S)SA -→---⎧-+⎨-→---⎩16手性反应注意事项● 手性试剂为高光学纯度试剂,光学稳定性好: ●● 手性待测物必须具备反应活性基团,如胺基(-NH2),醇基(-OH ),羧基(-COOH ),以保证与CDR 反应完全 ;● 生成的非对映体光学稳定,柱效要高,能有效检测。
17胺类对映异构体 ● 异硫氰酸酯(ITC )● 常用试剂有苯乙基异氰酸酯(PEIC )、2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖异硫氰酸酯(GITC ) 。
● 广泛应用于氨基酸及其衍生物、麻黄碱类、肾上腺素类、肾上腺素拮抗剂、儿茶胺类等药物的分离分析。
18GITC 衍生化分离胺类对映异构体 ● 反应原理C S NR NHR R 1R 22R 1●在三乙胺存在条件下进行,于乙腈、二氯甲烷或DMF中进行。
19色谱分离和前处理条件优化●流动相组成和流动相pH对拆分的影响●(1)降低甲醇含量改善分离度,保留时间增加;●(2)未衍生化的羧酸基对pH值比较敏感。
●衍生化时间及GITC用量的优化●(1)反应时间:10 min ~ 30 min(2)用量:过量两倍20手性拆分结果色谱条件:C18色谱柱,流动相:甲醇∶1%三乙基乙二氨四乙酸(TEAA 缓冲)(pH = 5.5),检测波长254 nm; 1 ml/min。
前处理:药物溶于含三乙胺乙腈水溶液,取50 μl该溶液,加入50 μl含2 g/L GITC的乙腈溶液,混合均匀后室温下放置10 min ~ 30 min,进样分析。
21拉贝洛尔的GITC衍生化23不同结构的药物衍生化策略●胺基类手性药物:运用异硫氰酸酯(ITC)、异氰酸酯(IC)类、手性酰氯、磺酰氯类试剂,将其衍生化为酰胺、氨基甲酸酯、脲、硫脲和磺酰胺。
●羧基类手性药物:运用手性胺类衍生化试剂,将其衍生化为酯和酰胺。
●醇类手性药物:运用手性酰氯、磺酰氯类试剂,将其衍生化成酯。
烯类手性药物:衍生化成水性铂复合物。
24手性固定相法(CSP)●原理:将手性试剂化学键合到固定相上与药物对映体反应形成暂时的非对映体对复合物,这种固定相称作CSP。
●在CSP表面所形成的非对映体对,可根据其稳定常数不同而获得分离。
(三点结合模型)25“三点相互作用”●对映体和手性固定相之间,至少需要三个同时发生的分子间相互作用力起作用,而其中至少有一个必须是立体化学相互作用。
●手性选择剂与对映体中的一种进入一个与立体相关的三点相互作用的稳定状态,而另一对映体则只能两点作用形成不稳定状态。
●这种稳定性相差越大,则相互分离的可能性就越大。
26“三点相互作用”●对映体和手性固定相之间,至少需要三个同时发生的分子间相互作用力起作用,而其中至少有一个必须是立体化学相互作用。
●手性选择剂与对映体中的一种进入一个与立体相关的三点相互作用的稳定状态,而另一对映体则只能两点作用形成不稳定状态。
●这种稳定性相差越大,则相互分离的可能性就越大。
Y 为S 性手性选择剂,X 为外消旋体 27 28蛋白质类手性固定相将蛋白质固定到硅胶上,利用蛋白质分子结构中的L-氨基酸提供手性作用位点与手性药物对映体产生不同的氢键作用、静电作用、疏水作用、离子对作用等达到手性拆分。
常用流动相为磷酸盐缓冲液(pH 4 ~ 7),流动相中可加入不超过5%有机溶剂。
29分类●白蛋白柱(HSA、BSA )●糖蛋白柱:α1-酸糖蛋白柱(α1- AGP )酶类30α1-酸糖蛋白柱拆分西酞普兰对映异构体色谱柱为Chiral-AGP 柱(150 mm×4.0 mm,5 μm)柱温为25℃;流动相为50 mmol/L醋酸铵缓冲液(乙酸调pH至4.1),流速0.8 ml/min;检测波长为240 nm。
31多糖及其衍生物类手性固定相● 纤维素的单体D-葡萄糖以β-1,4-糖苷键相连而成的线型聚合物,将羟基衍生化 成酯以增强手性识别能力。
● 由于水和卤代烃都能溶解纤维素及其衍生物,故流动相大都为正相色谱系统,使用极性较小的非卤代烃有机溶剂。
O***O OHOHO**OHO***OH OHO **OH O***OHOHO**OHO***OH OHO**OH32纤维素固定相手性识别作用手性中心附近带有羰基的外消旋化合物与纤维素衍生物通过偶极-偶极作用,带有羧基、羟基或氨基的外消旋化合物通过氢键发生手性识别作用。
33●直链淀粉是D-葡萄糖以α-1,4-糖苷键相连而成的线型聚合物,和纤维素的手性识别能力的不同,主要在于其葡萄糖单元的构象差异。
34最常用的多糖型手性色谱柱(Daicel ,大赛璐)●淀粉柱:Chiralpak AD、Chiralpak AS、●纤维素柱:Chiralcel OD、Chiralcel OJnsilica-gel35被用于分离布洛芬、氯胺酮等药物,以及在手性中心含有一个大的取代基的化合物36Chiralcel OD固定相分离美托洛尔对映体37●Chiralcel® OD-H手性色谱柱(4.6 mm×250 mm),●流动相为正己烷-异丙醇-二乙胺(65∶35∶0.1);流速为0.5ml/min;紫外检测波长为259 nm;●柱温为25℃;进样体积为20 μl。
38●环糊精类(包容色谱)●D-葡萄糖单元通过α-1,4-糖苷键连接的环状分子结构,其疏水性内腔的包容作用和腔外上的羟基与药物对映体的氢键作用是手性识别的基础。
有α、β、γ三种类型,其中β环糊精及其衍生物应用范围最为广泛。
39环糊精手性固定相改性●环糊精键合固定相机械强度差、不能在高压下使用,用氨基、氨基甲酸酯等改性,形成疏水相互作用或π-π相互作用、偶极-偶极叠合作用,扩大了手性拆分的能力,又称多模式手性固定相(multimodal CSP,MMCSP)。
4041手性流动相添加法(CMPA)●将手性试剂加到流动相中,利用下面两种方式实现拆分:①流动相中手性试剂与对映体形成非对映体配合物,在固定相中的保留时间和分配不同而得到拆分;②手性试剂吸附在柱上形成动态的手性固定相,对映异构体与之作用不同而得到拆分。
42环糊精手性固定相与流动相●流动相添加环糊精法拆分时,与流动相中环糊精包合越好的对映体,随流动相较快地被洗脱;●环糊精固定相拆分时,与固定相中环糊精包合越好的对映体易被固定相所保留,较慢地被洗脱。
●以环糊精流动相分离对映体的洗脱顺序与环糊精固定相拆分的顺序刚好相反。
43影响分离的因素有机溶剂:用量越少,保留时间越长,但是过高,则环糊精溶解度降低,显著影响对映体的分离度。
pH值酸度增大,有利于碱性药物的分离,对于酸性药物影响无规律性。
44●分子结构●(1)在其手性原子上均具有两个环状取代基或环内含有手性碳原子,而不能拆分的化合物在其手性碳原子上只含有一个环状取代基。
●(2)空间效应对手性识别的影响更为显著。
(3)环糊精浓度增加有利于手性分离,但β-环糊精溶解度受限制。
45手性流动相HPLC法拆分萘普生对映体H3CO* CH3优化的流动相添加剂:(a) L-脯氨酸、(b) β-环糊精(β-CD)、(e)羟丙基-β-环糊精(HP-β-CD)(c、d)甲基-β-环糊精(Me-β-CD)46●流动相体系:乙醇∶水相(不同手性添加剂,1%三乙胺水溶液,pH为4.4);HP-β-CD比甲基-β-CD拆分效果好47色谱分离和前处理条件优化●有机相对拆分的影响●(1)相比于乙腈,乙醇可以使保留时间缩短;●(2)随着乙醇体积分数的逐渐增大,其容量因子呈逐步减小的趋势,分离因子和分离度也减小。