QA-MS-018-1 包装材料抽检与
- 格式:doc
- 大小:77.00 KB
- 文档页数:3

目的建立通脉颗粒制剂工艺规程,使产品生产规范化,标准化,保证生产的产品质量稳定、均一和有效。
范围适用于通脉颗粒制剂生产全过程。
责任主管总经理、质量管理部部长、生产技术部部长、固体制剂车间主任、技术员标准依据《中华人民共和国药典》2000年版一部卫生部药品标准WS3-B-0824-91工艺规程的编制及管理规程(SMP.QA-GJ-01)内容1 产品概述1.1 品名:通脉颗粒汉语拼音:Tongmai Keli1.2 剂型:颗粒剂1.3 性状:本品为棕黄色的颗粒;气微,味甜、微苦。
1.4 功能与主治:活血通脉。
用于缺血性心脑血管疾病,动脉硬化,脑血栓,脑缺血,冠心病,心绞痛。
1.5 用法与用量:口服,一次10g,一日2~3次。
1.6 规格:每袋装10g。
1.7 贮藏:密封。
1.8 有效期:三年1.9 批准文号:国药准字1.10 包装规格:10g×8袋×108盒2.1 处方2.2 处方依据:卫生部药品标准WS3-B-0824-913 生产工艺流程图见第3页通脉颗粒生产工艺流程及环境区域划分示意图4 通脉颗粒生产质量控制要点5 制剂过程及工艺条件5.1 蔗糖粉碎将检验合格的蔗糖,投入涡轮自冷式粉碎机中,粉碎,过80目筛,执行“TF-160B型涡轮自冷式粉粹机标准操作规程”(SOP.SJ-SZ-35),装入不锈钢桶中。
送入存料间,称重,挂上标志。
5.2 制粒:按每批2万袋从提取中间站领料,二人复核,准确称量1/4配方量的清膏,糖粉投入槽型混合机中,执行“CH200A槽型混合机标准操作规程”(SOP.SJ-SZ-38),混合,待色泽达到均匀一致,将软材投入整粒机中,执行“KZ-180快速粉碎整粒机标准操作规程”(SOP.SJ-SZ-65),制得大小均匀颗粒。
5.3 干燥:将湿颗粒投入高效沸腾干燥机的沸腾器中,执行“GFG-120高效沸腾干燥机标准操作规程”(SOP.SJ-SZ-40),温度60℃,干燥30分钟,下料。


文件制修订记录1、抽样方案按GB/T2828.1-2012 (AQL:主0.65 次1.0)抽样标准进行随机抽检。
13 2、检验依据检验卡、包材料检验规范3、检验流程仓库开立送检单至IQC,IQC接送检单后应对产品标识单品名,型号及判定状态进行初步核对,若出现品名,型号或不合格产品直接做退货处理(外购产品除外),并如实对检验状况进行记录,开立品质异常通知单至上级部门进行确认并交送各相关部门进行处理。
4、环保检查若有需要,供方应提供具有权威性检测机构出示的有效期内的环保资料证书。
5、外观检验目视检查是否有如下不良现象:A、主要缺点:材料错误、结构不符、潮湿、油污、裂纹、多料、缺损、偏芯、变形、脱层、加工损坏、印刷错误、压痕、杂色、收缩、瓦绫稀疏、齿状、毛边等不良现象。
B、次要缺点:颜色稍有差异等轻微外观不良现象。
6、尺寸检验参照检验卡抽取数量,对来料厚度尺寸,长、宽、高尺寸进行测量,检查是否符合技术要求,并将测量结果记录于IQC检验记录表。
7、负荷检查A、外包装箱粘贴成型后平面应能够承受75㎏重量不变形,反折90°3次不得出现裂痕、破损。
B、内包装箱粘贴成型后平面应能够承受10㎏重量不变形,反折90°3次不得出现裂痕、破损。
C、包装塑管应能够承受10㎏重量不变形,扳动幅度状不得出现发白、断裂。
D、吸塑盒应能够承受3㎏重量不变形,反折90°3次不得出现裂痕、破损。
8、组装试验抽取相互配合包材进行试装验证(试装必须将所有包材以正常方式进行装配),组装后各相互包装盒/管不得出现挤压变形。
9、判定、处理依据检验卡对检验结果参照主次缺点允收水准接收值进行综合判定,完全符合要求则在产品识别标识单判定为合格并签名确认,若产品状况出现任一条件不相符超过主次缺点允收水准接收值,应判定为不合格处理,描述不合格原因并附不合格样品装订于产品识别标识单左上角,加盖红色NG印章。
随货移至不合格品区域,立即开立品质异常通知单至上级领导进行审批,并将审批后异常通知单分发相关单位办理退货处理、如实对送检单进行判定返送仓库,并对各项检验状况记录于IQC检验记录表报上级审批后存档。

2023年XX省危险化学品包装物容器产品质量监督抽查实施细则(补充)1 抽样方法以随机抽样的方式抽取检验样品和备用样品。
具体产品单元抽样数量见表1。
表1 抽样数量2 抽查产品名称及执行标准危险化学品包装物容器产品主要分为以下几种,各产品执行标准见下表。
表2 产品执行标准3 检验依据表3 钢质手提罐检验依据(产品标准BB/T0064-2013)重要程度分级:A类-极重要质量项目,B类-重要质量项目,C类-一般质量项目执行企业标准、团体标准、地方标准的产品,检验项目参照上述内容执行。
凡是注日期的文件,其随后所有的修改单(不包括勘误的内容)或修订版不适用于本细则。
凡是不注日期的文件,其最新版本适用于本细则。
依照有关规定或产品适用标准,需要检测的其他项目,可视情况进行调整。
4 判定规则4.1依据标准BB/T 0064-2013《包装容器钢质手提罐》现行有效的企业标准、团体标准、地方标准及产品明示质量要求4.2判定原则经检验,检验项目全部合格,判定为被抽查产品所检项目未发现不合格;检验项目中任一项或一项以上不合格,判定为被抽查产品不合格。
若被检产品明示的质量要求高于本细则中检验项目依据的标准要求时,应按被检产品明示的质量要求判定。
若被检产品明示的质量要求低于本细则中检验项目依据的强制性标准要求时,应按照强制性标准要求判定。
若被检产品明示的质量要求低于或包含本细则中检验项目依据的推荐性标准要求时,应以被检产品明示的质量要求判定。
若被检产品明示的质量要求缺少本细则中检验项目依据的强制性标准要求时,应按照强制性标准要求判定。
若被检产品明示的质量要求缺少本细则中检验项目依据的推荐性标准要求时,该项目不参与判定。
5 附则本细则是《XX省市场监督管理局关于发布第一批省级产品质量监督抽查实施细则的公告》(2023年4月3日)中《2023年XX省危险化学品包装物容器产品质量监督抽查实施细则》的补充细则,可同时执行。

Q E O M S 03第三层次文件
1.目的
确保我司包材来料检验能够符合我司及客户(IKEA )品质要求。
2.范围
适用于包装材料的检验。
【白盒﹑内衬﹑外箱﹑PET 盖、中包、托盘、封箱胶带、不干胶條碼﹑PE 袋﹑隔板(层垫)﹑打包带、护角等。
】 3.名词定义 无 4.权责
IQC 负责按此规范作业. 5.作业程序
5.1检验方法如下表
Q E O M S 03第三层次文件
5.3.1外来文件、图纸、检验标准
5.3.2供应商出具的检验报告或自我声明 5.3.3样品或相关限度样品 5.3.4到供应商处验证
5.4检验时间特殊时机处理
一般来料须在二日内完成检验,急件来料(仓库已无库存,生产线因够4小时使用)在4小时内完成检验,特急件来料(生产即将停线,等待来料投入)在2小时内完成检验,特殊情况即未经检验上线须经品管部经理同意后可首发20%物料上线生产,并告知IQPC 跟踪品质,剩余的80% IQC 必须马上验办,急件来料和特急件来料由计划在来料时立即通知品管人员处理. 5.5检验项目与方法: 5.5.1外观:目检。
5.5.2用卷尺,直尺测量、电子称、厚度仪、吸水性测试仪、扭力扳手等。
5.5.3颜色:目检。
5.6 公差依据:
5.6.1:按图面之公差.
5.6.2:
6.
6.1 IQC来料检验规程
7.质量记录
7.1 ABT-QEONMS04-QR802001来料检验报告
件
文
次
层
三
第
3
S
M
O
E
Q。

包裹层无纺布质量标准生效日期:文件批准日期编制部门:质量部修订记录1.目的规范包裹层无纺布材料质量检验操作,对包裹层无纺布材料进货质量进行控制,确保质量符合生产要求。
2.范围本标准规定了纸尿裤包裹层无纺布的技术要求,检验方法,检验规则及相关记录表单。
3.职责:公司研发部负责修订本标准,检验员依据本标准进行操作。
4.内容4.1.外观要求能按正常批次检测,有发现不合格批次后,需要验证2批合格后,才能转为正常批次检测。
4.4.安全要求害安全的问题,不做判定,直接退货。
5.抽样方案5.1批的组成:以一次交货数量为一批,交收检验样本单位为件,同一供应商,同一种类或同一等级组合成一批。
5.2抽样样本量:(除"重量"项目)按下表进行抽样,外观要求和尺寸要求(气味除外)按AQL=6.5进行判定,性能要求和安全要求按AQL=4.0进行判定。
备注:采用GB/T 2828.1 正常检查一次抽样方案检查水平S-26. 相关文件附录1:定量检测方法1、特殊仪器:电子天平(感量为0.01g ),0.01m 2圆盘取样器2、操作方法:①、将向折叠平整地铺放在裁样台上,量取该材料的宽度(b②、b ≥140mm 时折叠成3层使用圆盘取样器裁取,b<140mm 时折叠成5层沿纵向切取成1m 长的试样;③、将试样放置于电子天平上称量。
④.、试验结果计算:依下式计算每个试样的定量,精确至0.1g 。
3、计算结果:定量=M/S*P(g/m 2);式中M —试样的质量(g );S —试样的面积(m 2);b ≥140mm 时,S 取值0.01;b<140mm 时,S 取值为b (材料宽度值);P —层数 附录2:拉断力、延伸率测试方法1、仪器:CMT6102型拉力试验机 2 取样 2.2 测样数量:纵向5条,横向5条; 3 操作方法① 依次打开气动装置、拉力机、拉力机电脑、实验软件。
② 选择操作软件预先设定实验条件的“实验方案”。
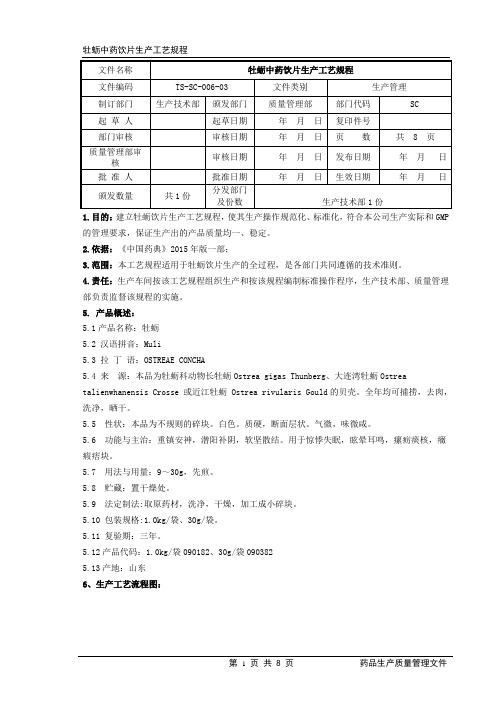
1.目的:建立牡蛎饮片生产工艺规程,使其生产操作规范化、标准化,符合本公司生产实际和GMP 的管理要求,保证生产出的产品质量均一、稳定。
2.依据:《中国药典》2015年版一部;3.范围:本工艺规程适用于牡蛎饮片生产的全过程,是各部门共同遵循的技术准则。
4.责任:生产车间按该工艺规程组织生产和按该规程编制标准操作程序,生产技术部、质量管理部负责监督该规程的实施。
5. 产品概述:5.1产品名称:牡蛎5.2 汉语拼音:Muli5.3 拉丁语:OSTREAE CONCHA5.4 来源:本品为牡蛎科动物长牡蛎Ostrea gigas Thunberg、大连湾牡蛎Ostrea talienwhanensis Crosse 或近江牡蛎 Ostrea rivularis Gould的贝壳。
全年均可捕捞,去肉,洗净,晒干。
5.5 性状:本品为不规则的碎块。
白色。
质硬,断面层状。
气微,味微咸。
5.6 功能与主治:重镇安神,潜阳补阴,软坚散结。
用于惊悸失眠,眩晕耳鸣,瘰疬痰核,癥瘕痞块。
5.7 用法与用量:9~30g,先煎。
5.8 贮藏:置干燥处。
5.9 法定制法:取原药材,洗净,干燥,加工成小碎块。
5.10 包装规格:1.0kg/袋、30g/袋。
5.11 复验期:三年。
5.12产品代码:1.0kg/袋090182、30g/袋0903825.13产地:山东6、生产工艺流程图:工艺流程线★---关键工艺控制点主要质量控制点7、操作过程及工艺条件7.1 批量及投料量:按实际生产确定批量和投料量7.2操作过程及工艺条件7.2.1炮制过程7.2.1.1领料:由净选工序的操作员按批生产指令领取规定的数量,填写领料单,核对原料物料编码,确保使用的牡蛎原料为一批。
7.2.1.2净选:按照《挑选工序标准操作规程》(SOP-SCGW-004)进行操作。
将牡蛎原药材置操作间内拣选台上,去净杂质等不符合要求的部分。
置周转箱内,转入下一道工序。

适用范围:本标准适用于印刷、复合(含淋膜复合)用普通塑料薄膜入仓前的检验。
铝箔AL 和聚酰胺薄膜(BOPA)参照2.2规定的内容。
标准内容:1.抽样标准及方法:1.1以该进货批次同种规格,同种材质的材料总数的10%抽验。
1.2拆开包装后,抽去膜卷表面1-2圈后取约1米长作为待测样品。
2.检验项目及方法:2.1印刷、贴合用普通塑料薄膜(PET、BOPP、CPP、LDPE)入仓前检验项目及方法2.2铝箔AL和BOPA薄膜因其特殊性,进料检验时不得开启原包装。
这两种材料进料验证时主要检查供方产品标识、合格证和供方检验报告的正确性和完整性。
AL 和BOPA的厚度、宽度、电晕处理值在原料上机使用拆除包装时由品管取样测试验证并做相应判定和记录。
3.批次检验结果与判定:上述检验指标全部合格才判定该抽样批次合格。
若有一项不合格则判为不合格。
但是某些不致影响材料本身应该具有的性能或不致影响最终产品的性能的缺陷,可以报上级视情形考虑降级使用。
4.记录与区分:4.1所有检验数据及判定结果,填入《进料检验记录》并交品管部主管确认。
4.2检验合格的材料作合格标识,交仓库于备料区区分摆放。
4.3遇有不合格物料,贴不合格标识,填写《不合格原料报告处理单》交相关部门领导确认后与供应商联络处理。
4.4检验员及时对检验合格和不合格的物料作出明确、固定的标识,并通知仓库按区域摆放。
附表1普通型双向拉伸聚丙烯薄膜1.外观应符合表1规定2.1薄膜宽度允差±2mm2.2厚度偏差、厚度平均偏差应符合表2规定符合GB9688之规定,嗅觉应无异味。
附件2热封型双向拉伸聚丙烯薄膜2.尺寸偏差4.卫生性能符合GB9688之规定,嗅觉应无异味。
附件3消光型双向拉伸聚丙烯薄膜1.外观应符合表1规定2.1薄膜宽度允差±2mm。
2.2厚度偏差、厚度平均偏差应符合表2规定3.物理机械性能应符合表4规定符合GB9688之规定,嗅觉应无异味附件4 珠光型双向拉伸聚丙烯薄膜2.1薄膜宽度允差±2mm。
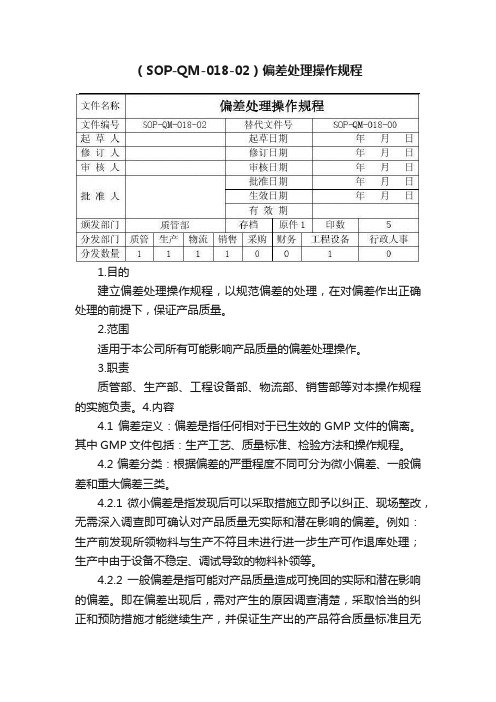
(SOP-QM-018-02)偏差处理操作规程1.目的建立偏差处理操作规程,以规范偏差的处理,在对偏差作出正确处理的前提下,保证产品质量。
2.范围适用于本公司所有可能影响产品质量的偏差处理操作。
3.职责质管部、生产部、工程设备部、物流部、销售部等对本操作规程的实施负责。
4.内容4.1 偏差定义:偏差是指任何相对于已生效的GMP文件的偏离。
其中GMP文件包括:生产工艺、质量标准、检验方法和操作规程。
4.2 偏差分类:根据偏差的严重程度不同可分为微小偏差、一般偏差和重大偏差三类。
4.2.1 微小偏差是指发现后可以采取措施立即予以纠正、现场整改,无需深入调查即可确认对产品质量无实际和潜在影响的偏差。
例如:生产前发现所领物料与生产不符且未进行进一步生产可作退库处理;生产中由于设备不稳定、调试导致的物料补领等。
4.2.2 一般偏差是指可能对产品质量造成可挽回的实际和潜在影响的偏差。
即在偏差出现后,需对产生的原因调查清楚,采取恰当的纠正和预防措施才能继续生产,并保证生产出的产品符合质量标准且无潜在质量风险。
例如:设备故障、损坏、清场不合格等。
4.2.3 重大偏差指已经或可能对产品质量造成不可挽回的实际和潜在影响的偏差。
偏差出现后,已明显对产品质量产生了影响,需对其重新处理或销毁处理等或者当时没有发现产品的质量发生变化,但产品质量在贮藏过程中可能存在隐患,对该产品进行重点留样及稳定性考察并可能采取产品召回等。
例如:关键参数偏离、测试结果未达到质量标准或超过警戒水平、混药、混批、包装材料混淆等。
4.3 偏差的处理方式:根据偏差的严重程度不同偏差的处理方式也不相同。
4.3.1 微小偏差的处理方式:微小偏差主要由发现人员采取应急措施并汇报工艺员和QA,工艺员负责对应急措施进行复核并对产生原因进行调查、提出纠正和预防措施。
QA对偏差的原因调查、处理措施和预防措施进行评估。
偏差发现部门经理和质管部部长对处理的过程和结果进行审核。

物料管理规范篇一:物料管理管理规程第四部分物料标准管理规程(一)亳州市豪门中药饮片目录1.物料代码管理规程 SMP-04-0012.物料进厂编号编制管理规程 SMP-04-0023.物料贮存养护管理规程SMP-04-0034.在库成品贮存养护管理规程 SMP-04-0045.不合格物料管理规程SMP-04-0056.不合格成品管理规程 SMP-04-0067.退库物料仓贮管理规程SMP-04-0078.定置管理规程 SMP-04-0089.成品包装、标签设计与印制管理规程SMP-04-009 10.标签发放使用及销毁管理规程 SMP-04-010 11.特殊药材管理规程SMP-04-011 12.物料超限额发放管理规程SMP-04-012 13.在库成品、物料申请复验管理规程 SMP-04-01314.仓贮状态标志管理规程 SMP-04-01415.库房养护设施管理规程 SMP-04-015 16.仓库清洁卫生及清场管理规程 SMP-04-01617.仓库安全管理规程SMP-04-01718.仓库盘存管理规程SMP-04-018编号:SMP-04-001-a原料代码表篇二:物料管理工作规范篇三:物料管理规程目的:对原材料进行规范化管理范围:所有进公司的原料、辅料、包装材料职责:仓库、质量检验部、供应部对此程序负责规程:定义:本规程物料是指:原辅料、中间体、成品及包装材料,成品单列文件管理。
1.原辅料及包装材料的采购1.1 供应部从质量部门获得的质量标准,作为寻求供货单位的依据。
供应部应注意从供货单位收集标准和方法,以便化验室进行比较和核对。
合格的采购员应掌握本公司主要物料的质量规格。
2. 工作程序2.1初步选择2.1.1供应部根据化验室提供的质量标准和供货单位能达到的标准进行对照,如达到标准或基本达到标准,则应了解供货单位的概况,包括供应商生产二证(营业执照、许可证)、产品工艺路线(流程图)、设备、工艺卫生状况、质量管理机构、工厂信誉等等,并根据这些基本情况对供货进行筛选。

文件目录:一.物料描述: (2)二.定性和定量的限度要求: (2)三.贮存条件和注意事项: (3)四.有效期或复验期: (3)五.取样、检验方法或相关规程编号: (3)六.变更记载及原因: (3)七.附录: (4)分发清单:质量部QA、质量受权人文件归档:质量管理保密等级:秘密□机密□绝密□一.物料描述:1. 物料名称:西林瓶2. 规格:2ml 5ml 15ml3.代码:57(2ml)58(5ml)59(15ml)4. 质量标准依据:参照《国家药用包装容器(材料)标准》(试行)YBB00292005-25. 生产厂:浙江新康药用玻璃有限公司二.定性和定量的限度要求:1.外观:应为无色透明;表面应光洁、平整,不应有明显的玻璃缺陷;任何部位不得有裂纹。
2.鉴别2.1.线性膨胀系数:应为5×10-6K-1(20-300℃)2.2.三氧化二硼的含量:不得小于8%(g/g)3.121℃颗粒法耐水性:应符合1级要求。
4.98℃颗粒法耐水性:应符合HGB1级要求。
5.内表面耐水性:应符合HC1级要求。
6.耐酸性:应符合1级要求。
7.耐碱性:应符合2级要求。
8.内应力:光程差不得过40nm/mm9.耐热性:不得破裂。
10.耐冷冻性:不得破裂。
11.砷浸出液:≤0.2mg/L12.锑浸出液:≤0.7 mg/L13.铅浸出液:≤0.2mg/L14.镉浸出液:≤0.25 mg/L15.垂直轴偏差:应符合附录表1规定的要求。
16. 尺寸:应符合附录表2规定的要求。
三.贮存条件和注意事项:无 四.有效期或复验期:无五.取样、检验方法或相关操作规程编号: 1. SOP-QA-042-01 包装材料取样操作细则 2.SOP-QA-038-01 包装材料检验操作细则 3. SOP-QA-016-01 取样,留样管理制度 4.六.变更记载及原因:附录表1 垂直轴偏差允许的最大值表2 规格尺寸。

-目建立克霉唑软膏的生产工艺规程。
的:范围:克霉唑软膏的生产。
职责:生产管理部、质量管理部、车间主任、班长、工艺员、操作工、QA。
规程:1.品名、剂型与处方依据1.1通用名称:克霉唑软膏汉语拼音:Kemeizuo Ruangao英文名:Clotrimazole Ointment1.2 剂型:乳膏剂1.3 处方与处方依据项的说明1.3.1处方(制成10000支量)克霉唑 3050g硬酯酸 10000g单硬酯酸甘油酯 4500 g白凡士林 8500g三乙醇胺 416.8g甘油 8500g羟苯乙酯 100 g玫瑰香精 50ml纯化水 65kg1.3.2 处方依据项说明:药品的生产批文:批准时间:质量标准编号: 2.工艺流程示意图:检验 →↓ →← 中间产品检验→← 成品检验3.生产工艺操作要求、工艺技术参数: 3.1 配制:3.1.1 配料操作工按照“软(乳)膏剂、凝胶剂配制岗位标准操作规程”SOP-MN/Z-(R )-001-00规定,在油相缸中加入处方量的硬酯酸、单硬酯酸甘油酯、白凡士林、液体石蜡加热至熔融,加入处方量的克霉唑,再加热至熔解并保温至94℃左右。
3.1.2配料操作工按照“软(乳)膏剂、凝胶剂配制岗位标准操作规程” SOP-MN/Z-(R )-001-00规定,另在水相缸中加入甘油、纯化水、三乙醇胺加热至沸腾,并在搅拌下加入用乙醇溶解的处方量羟苯乙酯,搅拌至均匀。
3.1.3先后将水相、油相抽入到真空乳化机内进行乳化,搅拌60分钟后,用冷却水冷却至48℃时,加入玫瑰香精,乳化后共搅拌120分钟。
3.2 中间品检验:检验室按“中间产品取样操作规程”规定,抽取配制好的膏体进行中间产品的检验。
检验合格后,发放“中间产品合格证”。
3.3 灌装、封尾:接到“中间产品合格证”后,车间按“软(乳)膏剂灌封岗位标准操作规程”SOP-MN/Z-(R )-002-00的要求,进入灌封工序。
封尾时随时抽检,注意剔除不合格品。

消毒供应中心包装材料及仪器设备标准中国国家标准GB/T4122.1—2008中规定,包装是:“为在流通过程中保护产品,方便贮运,促进销售,按一定技术方法而采用的容器、材料及辅助物等的总体名称。
也指为了达到上述目的而采用容器、材料和辅助物的过程中施加一定方法等的操作活动。
”包装的目的在于建立无菌屏障,确保器械物品在灭菌后预期的使用、运输和贮存等条件中保持无菌性,提供物理保护,并能无菌取用。
包装具体包括装配、包装、封包、注明标识等步骤。
医疗器械的具体性、预期的灭菌方法、预期的使用、有效期限、运输和贮存都对包装方法和材料选择带来影响。
包装材料指用于制造或密封无菌屏障系统或初包装的任何材料,必须能保证灭菌剂接触到器械,可提供微生物屏障。
任何待灭菌的器械物品必须加以包装,以保证器械在灭菌后至使用前的贮存期内保持无菌。
包装材料性质对保持无菌期限有直接影响,应选择尺寸合适的包装材料,以能将器械物品完全包裹为度,不能包裹太紧,以免影响空气的排出和灭菌剂的渗透。
医用包装材料应符合GB/T19633—2015或YY/T0698—2011要求的相关技术指标。
医院选择包装材料时,制造厂家应提供检测合格证书,医院感染管理部门和使用管理部门应进行审核。
消毒供应中心对购进的每批包装材料,应在入库前进行检查,并索要产品检测报告。
包装材料在功能性方面必须满足包装完整性、保护性便捷、洁净开启性3个条件,另外还要满足因灭菌方式不同的相适应性、材料的微生物阻隔性和无菌性的要求。
常用的包装材料有纺织材料、医用包装纸、无纺布、纸塑复合袋、硬质容器等。
一、纺织品/棉布纺织布是最传统、最简单的包装材料,目前在我国主要是棉布。
医院普遍用于使用频率高、周转快的手术器械的包装。
多年来的标准灭菌棉布均为每平方英寸(6.45cm2)140根纱的、未漂白的、双层厚度的棉布。
新棉布使用前应清洗。
重复使用的纺织包装材料每次使用后应清洗、消毒,使用前应在有灯的桌上检查,有破损的包装应禁止使用,不可以缝补后使用;使用前应去除棉绒。

医疗器械产品包装材料验证标准一、总则1 包装材料的要求参考依据:制定本规范参考了下列文件中的一些信息,但没有直接引用里面的条文。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注日期的引用文件,其最新版本适用于本标准.➢YY/T0681。
1➢YY/T0313。
用作制造的包装材料原料是原始材料,应有原料的来源,明确其历史和可追溯性,并受到控制,以确保成品始终能满足要求。
2 包装材料的设计必须在满足原定用途的条件下,既能够确保内包装材料的符合性,又把对使用者或患者的安全造成危害的可能性降低到最小程度.2。
1 包装材料与的相容性(即包装与医疗器材相互无不良影响):主要考虑的有:包装材料的安全性毒性的要求,拟包装的医疗器械的大小和形状,对物理和其它防护的要求,医疗器械对特殊危险例如辐射、湿气、机械性撞击,静电放射的敏感性。
2.2 包装材料与标识方式的相容性:标识方法必须对包装材料与采用的灭菌过程的相容性无不良影响,印刷或书写所采用的油墨不会转移到产品上,也不会和包装材料起反应而影响包装材料的效用,也不会变色而使标识变的模糊不清,对固定在包装材料表面的标识,其附着方式必须能耐受灭菌过程的使用及制造厂规定的贮存和运输条件。
3 包装材料能够提供对物理、化学和微生物的防护。
3.1包装材料在使用场所与使用者撕开包装取出使用时的要求相容性(例如无菌的开封)。
3.2 在使用条件下,在灭菌前、中、后,包装材料不可释放已知是有毒的,其数量足以对健康危害的物质。
3。
3无菌状态的保持:(即从其产品灭菌后,成为无菌之时起,直至规定的失效日期或使用时止),包装完整性及包装材料的微生物阻隔特性.3.4 材料的毒性检测.二、包装完整性试验1 试验目的对的包装系统,按照YY/T0681.1、YY/T0313 和“包装完整性试验方案”进行包装完整性验证,来评价包装系统的符合性。

文件分发号:__________ 审批过程版本历史记录一、目的:通过对供应商和服务商进行评估与选择,保证供应商和服务商具有满足本公司要求的能力。
二、范围:适用于评估为我公司提供生产用原辅料、包装材料的供应商和生产商、服务商。
三、责任:质量管理部、生产管理部,储运部,供应部。
四、程序:1、职责:1.1供应部:负责收集供应商资料,参与供应商和生产商、服务商的评估。
1.2质量管理部:负责组织对供应商评估,建立供应商和生产商、服务商档案,并于每年1月31日前完成本年度《合格供应商目录》,并制定相应的年度审计计划。
1.3生产管理部:负责参与供应商和生产商、服务商的评估。
1.4质量管理负责人:负责评估和批准合格供应商和生产商、服务商。
2、定义(供应商分为生产商和经销商)2.1供应商和生产商2.1.1供应商:直接提供商品及相应服务的企业及其分支机构、个体工商户,包括制造商、经销商和其他中介商。
2.1.2生产商:直接生产产品的制造商。
2.1.3生产商自己制造产品,而供应商不一定自己制造产品。
2.2供应商分类:2.2.1主要物料供应商:根据企业所生产的药品质量风险、物料用量以及物料对药品质量的影响程度确定我公司主要物料为:原料药的起始物料(如对氨基苯酚)、内包装材料和标签、印字外包材以及活性炭和冰醋酸。
主料物料的供应商为主要物料供应商。
2.2.2一般物料供应商:主要物料以外的物料供应商。
2.2.3服务商:包括可委托检验机构(强检仪表)、试剂供应商,关键设备设施供应商等。
服务商应有相关资质。
3、公司成立供应商质量体系评估工作小组,质量管理部设专人负责物料供应商质量评估和现场质量审计,由质量负责人参与并组织QA、生产管理部、供应部、生产车间等具有相关法规和专业知识的人员组成供应商评估小组。
审计人员应接受相关法规和相责人批准为合格供应商,列入合格供应商目录,我公司使用的原辅料、包装材料必须从列入合格供应商目录的供应厂家定点采购,需要采购的新原辅料或新开发的供应商应及时安排按规定程序进行质量评估。

供应商质量考核管理规定1.目的:针对供应商所供原料及产品在生产和使用过程中发生的质量问题,加强对供应商的质量整改和质量索赔,促进供应商采取补救和预防措施,持续改进质量,降低成本。
2. 范围:本规定适用于我公司供应商在供货期间的质量考核和质量索赔3.职责:质保部及生产车间是原材料及外购外协件不合格品报告的部门;质保部是对供应商的质量考核及对供应商采取纠正预防措施的相关部门;采购部是对供应商的考核联络部门;财务部是对供应商的质量考核进行帐务处理的部门。
4.质量问题分类A类质量问题:因供应件质量问题造成主机厂停线,产品功能失效,或引起客户重大质量索赔的。
B类质量问题:影响到产品功能、性能、可靠性、耐久性的质量问题,或严重影响顾客满意的。
C类质量问题:未出现影响产品的关键尺寸、功能、性能、可靠性、耐久性项目,仅在外观或非关键尺寸上有明显瑕疵的。
5 质量问题处置供应商在公司发生的质量问题按照附表1进行考核;造成生产车间停线的,按《供应商及时交付的管理规定》进行追加考核;影响客户产品交付的,按照附表1加倍考核。
在客户发生的质量问题,没有造成客户索赔的,按照附表1进行考核;造成客户索赔的追加客户的索赔金额。
按照附表1单项质量扣分在30分以上的,质保部发《整改通知单》。
供应商未及时提供的,按200元/天考核;措施无效,问题再次发生的,按1000~5000元进行考核。
生产线发现的不合格生产线发生重大、批量不合格时,采取紧急停用措施。
对车间不合格品进行标识隔离评审处置。
通知供应商进行现场解决,并采取相应措施,否则对责任供应商进行停用或取消配套资格。
客户发现的质量问题对供应商的考核客户处发生的重大的、批量不合格时,对供应商采取紧急停用措施。
因供应商产品连续两次在客户处发生重大质量事故的,取消供应商供货资格其他方面对供应商的考核质量保证能力在年度评定中没有达到规定要求的供应商,要求供应商限期整改,并实行限量供应。
2012年度质量目标:供应商零公里PPM值≤500,售后客户投诉次数为0供应商不具备必要的质量控制手段,或手段无效,也未在约定时间内整改到位。

*********科技有限公司GMP文件文件名称:留样管理规程文件编号:SMP-ZL-QA-018-00起草人日期年月日第 1 页,共 3 页审核人日期年月日分发号批准人日期年月日生效日期年月日颁发部门质量部分发部门质量部、生产部、采购部、研发部1 目的:建立留样管理规程,规范留样管理工作。
2 适用范围:适用于物料及产品的留样工作管理。
3 责任者:质量部4 管理内容:4.1 术语含义4.1.1 留样:企业按规定保存的、用于产品质量追溯或调查的物料、产品样品为留样。
用于产品稳定性考察的样品不属于留样。
4.2 质量部应按照原料、内包装材料、成品的贮存要求设立留样区域。
4.2.1 留样室内安装温度、相对湿度检测仪表。
留样室按照温度要求可分为常温(10~30℃)留样室及冷室(-5~-15℃,一般指冰箱冷藏区)。
湿度要求按品种要求而定,无特殊要求则为35%~70%。
每天上下午各检查一次留样室内各区的温湿度,并记录结果。
若阴凉室及冷室温湿度有偏差应采取措施纠正。
4.2.2 留样室由质量部安排QA管理。
4.2.3 留样室管理员负责检查、记录留样室的温度、相对湿度和留样样品的接收、保管、超过留样期限样品的销毁。
4.3 需留样的物料和产品4.3.1 每批原料和包装材料均应留样。
4.3.2 每批成品均应留样,如一批成品分次进行包装的,则每次包装应至少抽取一件最小包装对其留样。
4.4 留样数量原料、与产品直接接触的内包材及产品留样量至少为全检量的2倍。
外包材则按照取样数量进行留样,取样数量见《外包材抽检标准操作规程》。
成品的留样量为一次全检量的2~3倍。
4.5 样品的接收4.5.1 留样管理员在接收样品时,应检查所有容器是否均贴有标签,认真核对样品的名称、批号、数量等,填写相关留样台账。
对留样样品进行编号。
每批留样都应附有标签,标明样品名称、规格、批号,留样编号等。
排列整齐,易于识别。
4.5.2 留样编号的编制留样编号由一位大写英文字母及八位阿拉伯数字组成。

日 期AC:RE:合格不合格包装方式符合工艺或客户要求(塑封,打包带打包等)
无混装、漏装现象,外箱标识须全面正确(如无铅标识等)填写规范
包装箱上的标识必须清晰,正确,包装箱无破损;封箱胶带粘贴牢固,外观平整,无松脱,歪斜现象
2包材所有包材是否按要求摆放,规格正
确,数量齐全
是否按要求正确贴示流水号,商标,日期贴纸,PASSED 贴纸 等
外壳是否脏污,有无划痕,丝印是否正确清晰等
4功能
产品是否严格按照客户提供的测试要求执行
组装材料(PCB、外壳、螺丝、胶水、线材等)是否符合客户要求
连接方式(螺丝是否固定到位、胶水
是否多打少打漏打、连接线是否插好焊好)是否符合客户要求检验员:
审核:核准:QA检验报告(包装成品)
产品名称
检验依据型 号
班 别抽检数
抽检数检验结果不良记录1组装5机身外观3依据 GB/T2828.1-2012 一般检验水平的II级准执行,CR=0,MA=0.65,MI=2.5 进行判定抽检结果合格 确认人:
不合格(降级接收) 确认人:序号检验
项目检验内容
问题等级抽样数量彩盒/卡通箱。
1. 目的:制订本标准的目的是建立增加与减少包装材料抽样量的管理规程, 在进货质量不稳定时酌增抽样检验量, 而在连续检验质量稳定时, 减少取样检验量, 以降低检验人工成本。
2. 依据:国家药品监督管理局《药品生产质量管理规范》(1998年修订)第七十五条。
3. 范围:本标准适用于包装材料的抽样管理。
4. 责任:QA仓库检查员、包装材料检查员对本标准的实施负责。
5. 正文:
5.1. 包装材料的检验包括以下四类:
5.1.1. 正常检验:即按照检验规程进行正常量的抽样与检验。
5.1.2. 增量检验:在正常抽样量与检验项目的基础上,酌情增加抽样量或检验项目。
5.1.3. 减量检验:在正常抽样量与检验项目的基础上,酌情减少抽样量或对部分指标相对稳定的项目实行免检。
5.1.4. 终止检验:停止进货,待供应商对产品质量改进后,重新认证或更换供应商。
5.2. 各种检验类型的变更:
5.2.1. 正常检验→减量检验:在正常检验阶段, 在下列情况下可考虑酌减
抽样量或检验项目:
5.2.1.1. 连续5批检验合格;
5.2.1.2. 连续5批样品中不合格品总数未超过规定值;
5.2.1.3. 生产率稳定。
5.2.2. 正常检验→增量检验:正常检验时, 若连续5批中有2批不合格应酌增抽样量或检验项目。
5.2.3. 减量检验→正常检验:减量检验时, 在下列情况下应将抽样量或检验项目
增至正常:
5.2.3.1. 有1批不合格或有条件放行;
5.2.3.2. 供货不连续或长期停供;
5.2.3.3. 发生其它认为有必要增加抽样量或检验项目的情况。
5.2.4. 增量检验→正常检验:增量检验时, 若连续5批检验合格, 应考虑将抽样量或检验项目降至正常。
5.2.5. 增量检验→终止检验:增量检验时, 若连续5批增量检验,应终止检验, 通知供应商加以改进, 或更换供应商。
5.3. 转换程序:
5.3.1. 检验员检验完毕, 将检验结果登记于质量控制卡, 若连续检验结果符合转换条件, 报质量保证部经理核准后通知抽样员;
5.3.2. 抽样员在每批抽样时根据转换结果酌增或酌减抽样量;
5.3.3. 检验员根据转换结果酌增或酌减检验项目。
6. 附则:
6.1. 本标准附图0 幅,附表1 张。
6.2. 需要引用本标准的标准文件登记:
质量控制卡
编号:品名:规格:质量标准编号:。