第四章双原子分子的振动和转动
- 格式:pdf
- 大小:442.58 KB
- 文档页数:42

中国海洋大学光谱学课程大纲(理论课程)英文名称SPECTROSCOPY【开课单位】物理系【课程模块】专业知识【课程编号】【课程类别】选修【学时数】36 (理论28 实践8 )【学分数】2备注:课程模块为公共基础、通识教育、学科基础、专业知识或工作技能;课程类别为必修或选修。
一、课程描述本课程大纲根据2011年本科人才培养方案进行修订或制定。
(一)教学对象物理学专业、光信息科学与技术专业等具有原子物理专业基础的高年级本科生(二)教学目标及修读要求1、教学目标通过本课程的学习,使学生了解光谱学的原理和特点,知识认识物质结构同光谱之间的联系,进一步了解光谱学在现代生活、科技等领域中的应用。
通过了解光谱理论与技术在实际中的应用以及同物理、化学、光学等学科之间的联系,培养学生的综合运用知识能力、独立思考能力、学习意识和创新意识。
2、修读要求光谱学是一门多学科交叉的课程,主要研究光与物质之间的相互作用现象及作用机理,并被广泛应用于物质结构研究和分析。
课程介绍光谱学的基本概念、原理、有关计算与应用,在内容上包括原子光谱、分子光谱,振动光谱、转动光谱、电子光谱和散射光谱等。
通过光谱学基础课程的学习,进一步理解物质的能级结构与光谱的关系,掌握光谱学的基本概念与原理,对激光及激光光谱技术有一定的了解,了解光谱技术在实际中的应用;能够与本专业知识相结合,了解本专业知识同光谱学知识之间的相互促进关系,从而培养学生的创新意识及独立思考能力。
(三)先修课程(参照2011版人才培养方案中的课程名称,课程名称要准确)量子物理二、教学内容本课程主要讲述原子光谱和双原子分子光谱,具体内容及要求如下:绪论了解光谱学的研究对象和发展过程,它在新科学上的应用范围和发展。
第一章光谱概述1. 主要内容: 光的基本性质和光和物质相互作用的经典规律;发光和光谱物理机理;吸收、发射和散射光谱等基本概念;光谱学的应用。
2. 教学要求:了解光的基本性质和光和物质相互作用的经典规律;掌握发光和光谱物理机理;熟悉吸收、发射和散射光谱等基本概念;了解光谱学的广泛应用。

第四章振动光谱一、教学目的理解掌握震动光谱分析的基本理论,掌握红外光谱图的分析处理,了解红外光谱实验技术。
二、重点、难点重点:震动光谱分析的基本理论,红外光谱图的分析处理。
难点:震动光谱分析的基本理论。
三、教学手段多媒体教学四、学时分配4学时引言:●1900~1910年间,科布伦茨(W.W.C。
blentz)首先用红外光测量了一些有机物液体的吸收光谱而建立起一种新的分析方法——红外光谱法。
他发现分子中的一定原子群可以吸收特定的频率,这些特定的频率犹如人类的指纹,可以用来辨认分子中特定原子群的存在。
●它主要可以用作分子结构的基础研究和物质化学组成(物相)的分析(包括定性和定量)。
红外光谱法作分子结构的研究可以测定分子的键长、键角大小,并推断分子的立体构型,或根据所得的力常数,间接得知化学键的强弱,也可以从正则振动频率来计算热力学函数等。
●不过红外光谱法更多的用途是根据谱的吸收频率的位置和形状来判定本知物,并按其吸收的强度来测定它们的含量。
因此红外光谱法在目前已成为十分方便而有效的分析方法之一。
●红外光谱法应用得较多的是在有机化学领域,对无机化合物和矿物的红外鉴定开始较晚。
红外光谱法对测定矿物的结构或组分虽不如X射线衍射分析那么成熟,却也有其独特长处。
所谓振动光谱是指物质因受光的作用,引起分子或原子基团的振动,从而产生对光的吸收。
如果将透过物质的光辐射用单色器加以色散,使波长授长短依次排列,同时测量在不同波长处的辐射强度,得到的是吸收光谱。
如果用的光源是红外光波,即0.78~1000μm,就是红外吸收光谱。
如果用的是强单色光,例如激光,产生的是激光拉曼光谱。
本章主要介绍红外光谱的原理及其在无机非金属材料中的应用,对拉曼光谱只作简单的介绍。
红外光谱法就逐渐形成了一个极其有效而广泛的分析方法。
它主要可以用作分子结构的基础研究和物质化学组成(物相)的分(包括定性和定量)。
红外光谱法作分子结构研究可以测定分子的键长、键角大小,并推断分子的立体构型,或根据所得的力常数,间接得知化学键的强弱,也可以从正则振动频率来计算热力学函数等。


转动、振动很清楚的介绍见此书: Spectra Of Atoms And Molecules (Peter Bernat) 约化质量:BA B A M M M M +=μ 平衡键长:r e 或R e解离能:ZPE 0e +=D D (勿与下文D e 搞混,含义完全不同)双原子分子振动能量Fundamental (基频)的频率ν一般不按照频率来给出,而是通过ωe =h ν转换成能量后以cm -1为单位给出。
++-=≡+⎪⎭⎫ ⎝⎛++⎪⎭⎫ ⎝⎛+-⎪⎭⎫ ⎝⎛+=y x E y x E e e e vib e 3e 2e vib 814121)0(ZPE 212121)(ωωωωυωυωυυ υ:振动量子数 e ω:vibrational constant – first termx e ω:vibrational constant – second termy e ω:vibrational constant – third term双原子分子转动能量termdistortion al centrifigu 22rot ...)1()1()(++-+=J DJ J BJ J E J :转动量子数B :Rotational constant (cm -1)。
平衡键长时的值是B eD :Centrifigual distortion constant (cm -1),比B 小几个数量级。
有时也用D J 或D ele 表示。
平衡键长时的值是D e ,可通过Kratzer 关系得到2e 3e e /4ωB D =选律:∆J =±1对于多原子体系的振动问题,还要引入额外一个量子数K ,是J 在分子主轴上的投影。
双原子分子转-振能量B 和D 是依赖υ的。
考虑振动对转动能量的影响时...21...212118...)1()1()(e e 2e e e 222termdistortion al centrifigu 22rot +⎪⎭⎫ ⎝⎛++=+⎪⎭⎫ ⎝⎛++⎪⎭⎫ ⎝⎛+-==++-+=υβυγυαμπυυυυυD D B r h B J J D J J B J E e α:Vibration-rotation interaction constant 。

雙原子分子之振動古典力學中的彈簧振動可由牛頓力學描述:−ky = m (d 2y/dt 2)(1)其解為y = A cos ω t + B sin ω t (2)ω = (k/m)1/2 (3)振動頻率 ν =ω/2π; 常數A , B 可由起始條件決定。
在化學中雙原子分子之間的化學鍵也可視為一種彈簧,但由於原子是微觀的粒子,分子之振動不可以古典力學描述而須訴諸於量子力學。
雙原子分子振動之薛丁格方程式可寫成 ()()()x E x kx x dx d m ψψ2122222=+−! (4) 或()()0ψ2ψ22222=−+x kx m mE x dx d !! (5) x 為原子間之距離與平衡鍵長之差距,m 為reduced mass 。
這是一個二階homogeneous 常微分方程式,但由於係數不為常數,無法使用簡易的方式求解;我們將使用多項式法來處理這個問題。
首先,由於mk νπ21= (6) k = (2πν)2m (7)(5) 式可改寫為()()0ψα 2ψ22222= −+x x mE x dxd ! (8) 其中α = 2π ν m / ! (9)此時我們若直接以多項式求 ψ ,經驗告訴我們這並不易進行。
我們首先觀察到當x 很大時,(8)式可近似為()()0ψα ψ2222=−x x x dx d (10)若設2α2ψx e −= (11)2α2 αψx xe −−=′ (12) 2α222α22α222α)α( αψx x x e x e x e −−−≅−−=′′ (13)也就是說在x 很大時,(11)式為(10)式之解,或者說此時(11)式為(8)式之近似解。
因此,我們猜測(8)式中之波函數可寫成)( ψ2α2x f e x −=(14) 微分二次後得[]f x f f x f e x 222αααα2 ψ2+−′−′′=′′− (15) 將(13), (14) 式代入(8)中得0α2α2 2α2=−+′−′′−f mE f x f e x ! (16) 因此0α2α2 =−+′−′′f mE f x f ! (17) 我們現在嘗試以多項式法來解出fm m m x c x f ∑∞==)( (18) 10)(−∞=∑=′m m m x mc x f (19)20)1()(−∞=∑−=′′m m m x c m m x f (20)令n = m − 2此式可改寫為n n n x c n n x f ∑∞=+++=′′02)1)(2()((21) 將(18),(19)中之index 換成n 後與(21)一起代入(17)中我們得到 0α2 α2)1)(2(02002= −+−++∑∑∑∞=∞=∞=+n n n n n n n n n x c mE x c n x c n n ! (22)然而,若多項式恆為零則各項係數均需為零,因此0α2 α2)1)(2(22=−+−+++n n n c mE c n c n n ! (23)n n c n n mE n c )1)(2(/2α α222++−+=+! (24) 此為聯繫奇數項係數之間或偶數項係數之間關係的通式,但奇偶項之間並無關連。


《结构化学》课程教学大纲重点:H2+的线性变分法处理及其结果;双原子分子的基本理论(LCAO分子轨道法、价键法);分子轨道的构形、分类及其能级顺序;H2的海特勒-伦敦处理和价键理论。
难点:原子形成分子的规律;双原子分子的基本理论(LCAO分子轨道法、价键法);分子轨道的构形和能级顺序。
第四章分子对称性和分子点群(6学时)知识点:对称元素和对称操作;群论的基本知识;分子点群;群表示理论的要点。
重点: 对称元素和对称操作的概念;判别分子对称群的方法和有关应用;分子对称性与性质的关系。
难点: 判别分子对称群的方法;群表示理论的要点;分子对称性与性质的关系。
第五章多原子分子结构(6学时)知识点:杂化轨道理论;价层电子对互斥理论;多中心键和缺电子分子结构;离域 键和共轭分子结构;分子轨道的对称性和反应机理;配位化合物:价键理论、晶体场理论、配位场理论。
重点:杂化轨道理论及波函数、价电子对互斥理论的概念和应用;分子构型的判断;多中心键和缺电子分子结构。
共轭分子轨道求解的过程和能级计算;共轭分子分子图及应用;配位场理论。
难点:杂化轨道理论及波函数;价层电子对互斥理论分子构型的判断。
休克尔分子轨道法;分子图;分子轨道的对称性和反应机理。
第六章晶体结构基础(8学时)(1)几何结晶学知识点:晶体内部结构的空间点阵排列规律;晶体的对称性以及晶体宏观和微观对称类型(32种点群和230种空间群)。
晶体的晶面符号;根据晶体对称性或内部空间点阵的分布规律进行晶体分类(七个晶系和十四种空间点阵);晶体的特性;晶面角守恒定律。
重点:晶体内部结构的空间点阵排列规律;晶体的对称性,宏观和微观对称类型;根据晶体对称性情况或内部空间点阵的分布规律进行分类;晶面角守恒定律。
难点:晶体内部质点的空间点阵排列规律;晶体的对称性,晶体对称类型。
(2)晶体化学知识点:晶体中的价键,等径圆球和不等径圆球的密堆积;金属单质、非金属单质、合金的晶体结构;离子键和典型离子化合物、复杂离子化合物晶体结构的描述;鲍林规则和硅酸盐晶体的结构;某些三元化合物的晶体结构及近代晶体材料简介。

转动、振动很清楚的介绍见此书: Spectra Of Atoms And Molecules (Peter Bernat) 约化质量:BA B A M M M M +=μ 平衡键长:r e 或R e解离能:ZPE 0e +=D D (勿与下文D e 搞混,含义完全不同)双原子分子振动能量Fundamental (基频)的频率ν一般不按照频率来给出,而是通过ωe =h ν转换成能量后以cm -1为单位给出。
++-=≡+⎪⎭⎫ ⎝⎛++⎪⎭⎫ ⎝⎛+-⎪⎭⎫ ⎝⎛+=y x E y x E e e e vib e 3e 2e vib 814121)0(ZPE 212121)(ωωωωυωυωυυ υ:振动量子数 e ω:vibrational constant – first termx e ω:vibrational constant – second termy e ω:vibrational constant – third term双原子分子转动能量termdistortion al centrifigu 22rot ...)1()1()(++-+=J DJ J BJ J E J :转动量子数B :Rotational constant (cm -1)。
平衡键长时的值是B eD :Centrifigual distortion constant (cm -1),比B 小几个数量级。
有时也用D J 或D ele 表示。
平衡键长时的值是D e ,可通过Kratzer 关系得到2e 3e e /4ωB D =选律:∆J =±1对于多原子体系的振动问题,还要引入额外一个量子数K ,是J 在分子主轴上的投影。
双原子分子转-振能量B 和D 是依赖υ的。
考虑振动对转动能量的影响时...21...212118...)1()1()(e e 2e e e 222termdistortion al centrifigu 22rot +⎪⎭⎫ ⎝⎛++=+⎪⎭⎫ ⎝⎛++⎪⎭⎫ ⎝⎛+-==++-+=υβυγυαμπυυυυυD D B r h B J J D J J B J E e α:Vibration-rotation interaction constant 。
2组长:070601314组员:070601313070601315070601344070601345070601352第四章 双原子分子结构与性质1.简述 LCAO-MO 的三个基本原则,其依据是什么?由此可推出共价键应具有什么样的特征?答:1.(1)对称性一致(匹配)原则: φa = φs 而φb = φ pz 时, φs 和φ pz 在σˆ yz 的操作下对称性一致。
故 σˆ yz ⎰φs H ˆφ pz d τ = β s , pz ,所以, β s , pz ≠ 0 ,可以组合成分子轨道(2)最大重叠原则:在 α a 和α b 确定的条件下,要求 β 值越大越好,即要求 S ab 应尽可能的大(3)能量相近原则: 当α a = α b 时,可得 h = β ,c 1a = c 1b , c 1a =- c 1b ,能有效组合成分子轨道;2.共价键具有方向性。
2、以 H 2+为例,讨论共价键的本质。
答:下图给出了原子轨道等值线图。
在二核之间有较大几率振幅,没有节面,而在核间值则较小且存在节面。
从该图还可以看出,分子轨道不是原子轨道电子云的简单的加和,而是发生了波的叠加和强烈的干涉作用。
图 4.1 H + 的 ψ 1(a)和 ψ 2(b)的等值线图研究表明,采用 LCAO-MO 法处理 H 2+是成功的,反映了原子间形成共价键 的本质。
但由计算的得到的 Re=132pm ,De=170.8kJ/mol ,与实验测定值Re=106pm、De=269.0 kJ/mol 还有较大差别,要求精确解,还需改进。
所以上处理方法被称为简单分子轨道法。
当更精确的进行线性变分法处理,得到的最佳结果为Re=105.8pm、De=268.8 kJ/mol,十分接近H2+的实际状态。
成键后电子云向核和核间集中,被形象的称为电子桥。
通过以上讨论,我们看到,当二个原子相互接近时,由于原子轨道间的叠加,产生强烈的干涉作用,使核间电子密度增大。
2组长:070601314 组员:070601313070601315070601344070601345070601352第四章 双原子分子结构与性质1.简述 LCAO-MO 的三个基本原则,其依据是什么?由此可推出共价键应具有什么样的特征?答:1.(1)对称性一致(匹配)原则: φa = φs 而φb = φ pz 时, φs 和φ pz 在σˆ yz 的操作下对称性一致。
故 σˆ yz ⎰φs H ˆφ pz d τ = β s , pz ,所以, β s , pz ≠ 0 ,可以组合成分子轨道(2)最大重叠原则:在 α a 和α b 确定的条件下,要求 β 值越大越好,即要求 S ab 应尽可能的大(3)能量相近原则: 当α a = α b 时,可得 h = β ,c 1a = c 1b , c 1a =- c 1b ,能有效组合成分子轨道;2.共价键具有方向性。
2、以 H 2+为例,讨论共价键的本质。
答:下图给出了原子轨道等值线图。
在二核之间有较大几率振幅,没有节面,而在核间值则较小且存在节面。
从该图还可以看出,分子轨道不是原子轨道电 子云的简单的加和,而是发生了波的叠加和强烈的干涉作用。
图 4.1 H +的 ψ 1(a)和 ψ 2(b)的等值线图研究表明,采用 LCAO-MO 法处理 H 2+是成功的,反映了原子间形成共价键 的本质。
但由计算的得到的 Re=132pm ,De=170.8kJ/mol ,与实验测定值Re=106pm、De=269.0 kJ/mol 还有较大差别,要求精确解,还需改进。
所以上处理方法被称为简单分子轨道法。
当更精确的进行线性变分法处理,得到的最佳结果为Re=105.8pm、De=268.8 kJ/mol,十分接近H2+的实际状态。
成键后电子云向核和核间集中,被形象的称为电子桥。
通过以上讨论,我们看到,当二个原子相互接近时,由于原子轨道间的叠加,产生强烈的干涉作用,使核间电子密度增大。
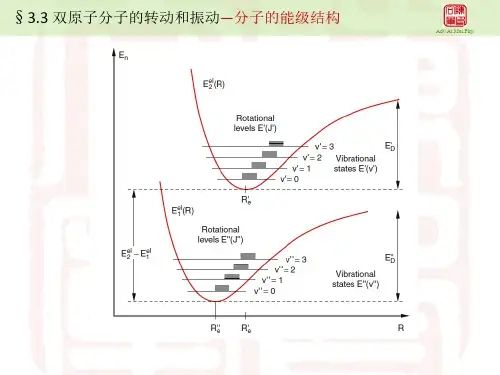
第四章 双原子分子的振动和转动§4-1 分子光谱概述㈠ 带状光谱对于原子而言,原子的能量是量子化的,只能取某些确定值。
例如,He 原子,在中心场近似下,各能级 是1s 2 、1s 2s 、1s 2p 、1s 3s 等。
当原子从一个状态变化到另一个状态时,伴随着电子的跃迁,将吸收或发射 光子, hv E = D 。
由于原子的能量只能取某些特定值,跃迁时吸收或发射的光的频率也只能是某些特定值, 反映在光谱上是一些分立的谱线。
因此,原子光谱是线状光谱。
分子的运动比原子复杂,不仅要考虑电子的运动,还要考虑核的运动。
分子内部运动总能量为rv e E E E E + + = 其中, E e 是电子的能量,它包括纯电子能量和核间排斥能(纯电子能量则包括电子的动能、电子间的排 斥能、电子与核之间的吸引能);E v 是核的振动能;E r 是分子转动能。
· 每个电子运动状态对应着一个电子能级,间隔约为120eV 。
· 每一电子运动状态下,有不同振动状态,每个振动状态对应着一个振动能级,能级间隔0.051eV 。
· 每个振动状态下,有不同的转动状态,每个转动状态对应着一个转动能级,能级间隔10 4 10 2 eV 。
¨ 分子吸收或发射光辐射时,单纯改变转动状态是可以的,产生转动光谱,位于微波和远红外区。
¨ 振动状态变化时,往往伴随着转动状态的变化,产生振动转动光谱,位于红外区。
¨ 电子状态变化时,往往伴随着振动、转动状态的变化,产生电子光谱,位于近红外、可见或紫外区。
由于这些能级非常靠近,谱线非常密集,在低分辨率的仪器下,不能将谱线分开,看上去象连续的谱带, 因此,分子光谱表现为带状光谱。
㈡ 核运动的处理以双原子分子为例。
首先处理电子运动,根据波恩奥本海默近似,假设核是固定的,elel NN el R U V H Y = Y + ) ( ) ˆ ( 或 elel el el R E H Y = Y ) ( ˆ el H ˆ 包含电子的动能、电子间的排斥能、电子与核之间的吸引能; NNV 是核间的排斥能。