慢性炎症性脱髓鞘性多发性神经病参考PPT
- 格式:ppt
- 大小:123.50 KB
- 文档页数:28



可治性罕见病—慢性炎性脱髓鞘性多发性神经根神经病一、疾病概述慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)是一类由免疫介导的获得性感觉运动神经脱髓鞘性多发性神经病。
1890年由Eichhorst首先描述,1982年由Dyck等正式命名为CIDP。
该病估计患病率为1/100000—9/100000。
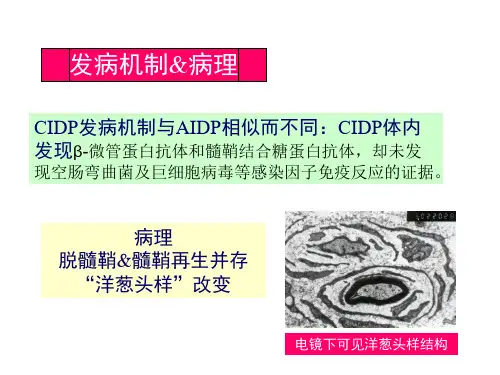
该病呈慢性进展或缓解复发,经典型主要表现为四肢近端和远端的无力和感觉障碍,可伴有脑脊液蛋白-细胞分离,神经电生理表现为运动和感觉神经的传导速度减慢、传导阻滞、远端潜伏期延长及波形离散,病理可见炎性细胞浸润、脱髓鞘、髓鞘再生并呈洋葱球样改变。
对糖皮质激素、免疫球蛋白和血浆置换等免疫调节治疗有效。
随着对本类疾病认识的深入,目前将CIDP分为以下几种亚型:经典型、运动型、感觉型、远端获得性脱髓鞘性对称性神经病(DADS)、多灶性获得性脱髓鞘性感觉运动神经病(Lewis-Sumner综合征)、局灶性CIDP、急性发作性CIDP 等[1]。
二、临床特征本病病程大于8周,经典型的主要临床特点为慢性进展或缓解复发的四肢近端和远端肌无力和感党障碍,四肢腱反射迟钝。
运动型以复发缓解的四肢肌无力为主要表现,无感觉受累,多见于20岁以下患者。
感觉型常首先表现为双下肢麻木,部分患者会在感觉症状出现数年后发展至运动功能受累。
DADS以远端对称感觉障碍为主要表现,部分患者也有运动功能障碍。
Lewis-Sumner综合征是一种多数单神经病,通常从上肢起病,多表现为抓握、手指外展无力,伴有感觉障碍。
局灶型则表现为局限在某个肢体或肢体某个区域的感觉和运动功能障碍,可以持续数年后逐渐向其他区域发展。
急性发作的CIDP表现为在8周内病情急性进展,在诊断上容易与急性炎性脱髓鞘性多发性神经根神经病(AIDP)相混淆。
本病脑脊液呈现蛋白-细胞分离,神经根丛MRI扫描可见水肿、萎缩和偶见的强化。