4-7 注射用无菌粉末
- 格式:ppt
- 大小:1.51 MB
- 文档页数:31


化学药品注射剂灭菌/无菌工艺研究及验证指导原则目录一、概述 (3)二、注射剂湿热灭菌工艺 (4)(一)湿热灭菌工艺的研究 (4)1.湿热灭菌工艺的确定依据 (4)2.微生物污染的监控 (7)(二)湿热灭菌工艺的验证 (9)1.物理确认 (9)2.生物学确认 (12)3.基于风险评估的验证方案设计 (15)三、注射剂无菌生产工艺 (15)(一)无菌生产工艺的研究 (15)1.除菌过滤工艺的研究 (16)2.无菌分装工艺的研究 (17)(二)无菌生产工艺的验证 (18)1.除菌过滤工艺验证 (18)2.无菌工艺模拟试验 (21)四、附件 (24)五、参考文献 (26)1一、概述2无菌药品是指法定药品标准中列有无菌检查项目的制3剂和原料药,一般包括注射剂、无菌原料药及滴眼剂等。
4从严格意义上讲,无菌药品应不含任何活的微生物,但由5于目前检验手段的局限性,绝对无菌的概念不能适用于对6整批产品的无菌性评价,因此目前所使用的“无菌”概念,7是概率意义上的“无菌”。
特定批次药品的无菌特性只能通8过该批药品中活微生物存在的概率低至某个可接受的水平,即无菌保证水平(Sterility Assurance Level, SAL)来表征,910而这种概率意义上的无菌需通过合理设计和全面验证的灭11菌/除菌工艺过程、良好的无菌保证体系以及在生产过程中12执行严格的药品生产质量管理规范(GMP)予以保证。
13本指导原则主要参考国内外相关技术指导原则和标准14起草制订,重点对注射剂常用的灭菌/无菌工艺,即湿热灭菌为主的终端灭菌工艺(terminal sterilizing process)和无1516菌生产工艺(aseptic processing)的研究和验证进行阐述,17旨在促进现阶段化学药品注射剂的研究和评价工作的开展。
本指导原则主要适用于无菌注射剂申请上市以及上市后变1819更等注册申报过程中对灭菌/无菌工艺进行的研究和验证20工作,相关仪器设备等的验证及常规再验证不包括在本指导原则的范围内。

不溶性微粒检查法(通则0903)培训试题及答案2018.08姓名:成绩:一、单选题(每题4分,共20分)1、光阻法仪器测量粒径范围为:。
(A)A、2~100μmB、2~100nmC、0.1~10μmD、50~200μm2、光阻法仪器检测微粒浓度为:。
(B)A、0~10000个/LB、0~10000个/mlC、0~20000个/mlD、0~15000个/ml3、光阻法仪器的校准周期为:个月。
(C)A、12B、9C、6D、34、微粒检查用水(光阻法)的要求:。
(B)A、≧10μm的不溶性微粒≦5粒/10ml≧25μm的不溶性微粒≦1粒/10mlB、≧10μm的不溶性微粒≦10粒/10ml≧25μm的不溶性微粒≦2粒/10mlC、≧10μm的不溶性微粒≦10粒/5ml≧25μm的不溶性微粒≦2粒/5mlD、≧10μm的不溶性微粒≦20粒/10ml≧25μm的不溶性微粒≦5粒/10ml5、标示装量≧100ml的大容量静脉用注射液(光阻法)的不溶性微粒结果判定:(A)。
A、≧10μm ≦25粒/ml≧25μm ≦3粒/mlB、≧10μm ≦12粒/ml≧25μm ≦2粒/mlC、≧10μm ≦25粒/5ml≧25μm ≦3粒/5mlD、≧10μm ≦25粒/10ml≧25μm ≦3粒/10ml二、多选题(每题4分,共20分)1、《中国药典》2015年版中不溶性微粒检查法包括:。
(AB)A、光阻法B、显微计数法C、电阻法D、沉降法2、光阻法仪器通常包括部分。
(ABC)A、取样器B、传感器C、数据处理器D、雾化器3、《中国药典》2015年版通则规定仪器使用前应对进行校准。
(BCD)A、波长准确度B、取样体积C、微粒计数D、传感器分辨率4、在光阻法测量时,影响测定结果的因素主要包括:。
(ABCD)A 、环境B 、检品取样方式、仪器设备C 、样品性质D 、样品稀释倍数5、显微计数法通常使用的装置包括: 。
(ABCD ) A 、洁净工作台 B 、显微镜 C 、微孔滤膜 D 、滤器、平皿三、判断题(每题 2分,共20 分)1、使用光阻法检测时,操作一般在洁净工作台,或符合要求的洁净实验室中进行,以确保无外来微粒引入,供试品溶液不被污染。

注射用冷冻干燥无菌粉末用溴化丁基橡胶塞本标准适用于直接与注射用冷冻干燥无菌粉末接触的溴化丁基橡胶塞的检验。
【外观】取本品数个,照附表检查法检查,应符合规定。
【鉴别】* (1)取本品适量剪成小颗粒,称取2.0g,置于30ml坩埚中,加碳酸氢钠2.0g均匀覆盖试样,置电炉上,缓缓加热至炭化,放冷,置高温炉300℃加热至完全灰化,取出,放冷,加水10ml使溶解,过滤,取滤液1.5ml,置于试管中,加硝酸酸化,加入硝酸银试液1滴,应产生淡黄色沉淀。
(2)除另有规定外,照包装材料红外分光光谱测定法(YBB00262004)第四法测定,应与对照图谱基本一致。
【穿刺落屑】取本品10个,照注射剂用胶塞、垫片穿刺落屑测定法(YBB00332004)第二法对照法测定,落屑数应不得过5粒。
【穿刺力】取本品10个,照注射剂用胶塞、垫片穿刺力测定法(YBB00322004)第二法测定,穿刺瓶塞所需的力均不得过10N。
【胶塞与容器密合性】取本品10个,置烧杯中,加水5分钟,取出,在70℃干燥1小时,备用。
另取10个与之配套的注射液瓶,加水至标示容量,用上述胶塞、垫片塞紧或封紧,再加上与之配套的铝盖或铝塑盖,压盖。
放入高压灭菌器中,121℃±2℃,保持30分钟,冷却至室温,放置24小时,将上述样品倒置,放入含有10%亚甲兰溶液的容器中,置于带抽气着装置的容器中,抽真空度为25kPa,维持30分钟,真空装置恢复至常压,再放置30分钟,取出,用水冲洗瓶外壁,观察,亚甲兰溶液不得渗入瓶内。
【自密封性】取胶塞与容器密合性项下样品,采用符合注射剂用胶塞、垫片穿刺力测定法(YBB00322004)第二法中注射针,向胶塞不同穿刺部位垂直刺穿胶塞,每个胶塞穿刺3次,每穿刺10次后更换注射针。
将上述样品倒置,放入含有10%亚甲兰溶液的容器中,置于带抽气着装置的容器中,抽真空度为25kPa,维持30分钟,真空装置恢复至常压,再放置30分钟,取出,用水冲洗瓶外壁,观察,亚甲兰溶液不得渗入瓶内。


1一、概述2对于临床使用过程中需复溶和/或稀释后使用的化学药3品注射剂,如注射用无菌粉末和注射用浓溶液等,需进行配4伍稳定性研究,考察药物在临床配制、存放和使用过程中质5量随时间的变化情况,为注射剂药品的配制、配制后药液的6存放条件和允许时限等提供依据。
本指导原则重点阐述化学7药品注射剂复溶和/或稀释配伍稳定性研究的试验样品、试验8设计、试验结果评估和说明书相关内容撰写等方面内容,为9研发和技术审评提供参考。
10本指导原则主要适用于化学药品注射剂上市申请与一11致性评价申请,不包括放射性药品。
12本指导原则仅代表药品监管机构当前的观点和认识,不13具有强制性的法律约束力。
随着科学研究进展,本指导原则14中的相关内容将不断完善与更新。
15二、总体考虑16根据药品特性与临床使用情况开展配伍稳定性研究。
上17市注册申报配伍稳定性试验的样品应具有代表性,并涵盖新18生产样品和近效期样品。
配伍稳定性药液的浓度应涵盖临床19使用中的最高和最低浓度,考察时间应不短于说明书中允许20时限,研究时应尽可能模拟药物临床使用中的实际情况。
由21于稀释后药物浓度降低可能导致杂质无法准确检出,或配伍22后可能产生新杂质,应注意评估分析方法的适用性。
23新药可根据药品特性、临床需要、临床使用条件(时间、24温度、光照等)等合理设计试验,在新药研发早期,可采用25小试样品或早期临床样品开展配伍稳定性研究,新药研发中26发生处方变更等情况时,可基于风险评估和需要重新开展配27伍稳定性研究,上市注册申报配伍稳定性试验样品应具有代28表性,对配伍稳定性中出现的新杂质应按杂质研究相关指导29原则进行归属研究,必要时进行安全性研究或提供其他安全30性依据。
根据研究结果在说明书中明确药品的配制方法、保31存条件和允许时限等相关内容。
32仿制药质量和疗效应与参比制剂一致。
应参照参比制剂33说明书进行临床配伍稳定性研究,参比制剂说明书中配伍相34关信息不明确的,建议参照新药要求开展配伍稳定性研究,35并与参比制剂进行对比研究。
![第四节粉针剂[专业类别]](https://uimg.taocdn.com/4489e1ffa417866fb94a8e2a.webp)



实验五 注射剂的制备一、实验目的1. 通过实验建立无菌概念,掌握无菌与灭菌制剂生产工艺中的关键操作。
2. 掌握注射剂生产的工艺过程和操作要点。
包括注射剂所用容器的处理、配液、滤过、灌封通气、灭菌等基本操作。
3. 熟悉注射剂成品质量检查标准和方法,包括安瓿的漏气检查、澄明度检查、pH值检查等基本操作,了解影响成品质量的因素。
4. 熟悉无菌操作室的洁净处理、空气灭菌等。
二、实验原理注射剂系指用药物制成的供注入体内的无菌溶液、乳状液和混悬液,以及供临用前配制成溶液或混悬液的无菌粉末。
注射剂起效迅速,剂量准确,特别是常作急救危重病人用的静脉滴注的输液。
由于注射剂直接注入体内,吸收快,所以对生产过程和质量控制,都要求极其严格。
注射剂的质量要求包括无菌、无热原、澄明度合格、使用安全、应无毒性和刺激性;注射液的 pH值应接近体液,一般控制在4-9范围内;凡大量静脉注射或滴注的输液,应调节渗透压与血浆渗透压相等或接近;稳定性合格,即在贮存期内稳定有效;含量合格;在水溶液中不稳定的药物,常制成注射用无菌粉末,以保证注射剂在贮存期内稳定、安全、有效。
生产灭菌制剂的厂房设施必须根据卫生部颁布的《药品生产质量管理规范》(GMP)的原则设置,厂房必须按生产工艺和产品质量的要求划分洁净级别。
一般可分别一般生产区、控制区、洁净区。
一般生产区指无洁净度要求的生产或辅助房间。
控制区是指对空气洁净度有一定要求的生产或辅助房间。
洁净区是指有较高洁净度和菌落数要求的生产房间。
房间设计的布局要合理,人流物流要严格分开。
洁净级别要求高的厂房对相邻的洁净级别低的厂房一般呈相对正压。
生产车间及各岗位操作区,均应按生产和洁净级别的要求进行清洁、消毒。
洁净厂房内空气的尘粒数和活微生物数应符合规定,温度和相对湿度应与其生产及工艺要求相适应。
注射剂的灭菌方法,应根据灭菌的药物及其制剂的稳定性进行选择。
热压灭菌法是制备注射剂和滴眼剂最常使用的方法。
热压灭菌器系受压容器,使用时要谨慎,应遵守正规的操作规程,以免发生事故。
1.增溶剂、助溶剂、潜溶剂的异同潜溶剂:在混淆溶剂中各溶剂在某一比率时,药物的溶解度比在各纯真溶剂中的溶解度大,并且出现极大值,这类现象称为潜溶,这类溶剂称为潜溶剂。
潜溶剂能提升药物溶解度的体制一般以为是两种溶剂间发生氢键缔合,改变了混淆溶剂的极性,即降低了溶剂的介电常数,进而有益于难溶性药物的溶解。
助溶剂:难溶性药物与加入的第三种物质在溶剂中形成可溶性络合物、复盐或缔合物等,以增添药物在溶剂中的溶解度,这第三种物质称为助溶剂。
增溶剂:指某些表面活性剂增大难溶性药物的溶解度的作用。
拥有增溶能力的表面活性剂称增溶剂。
原理:表面活性剂在水中形成胶束的结果。
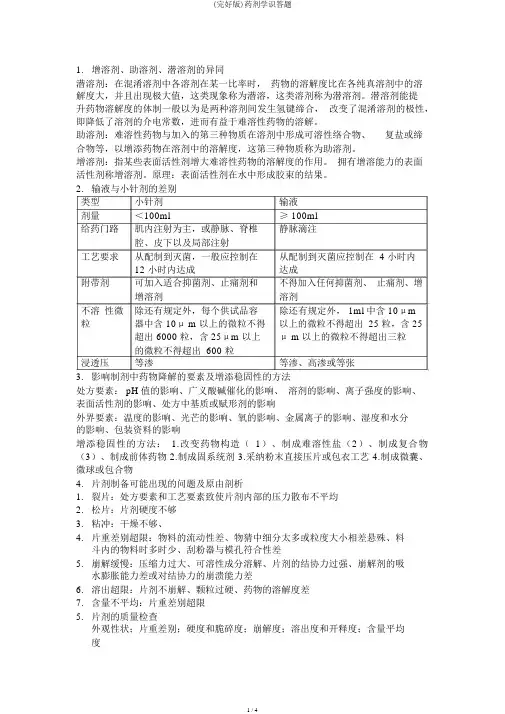
2.输液与小针剂的差别类型小针剂输液剂量<100ml≥ 100ml给药门路肌内注射为主,或静脉、脊椎静脉滴注腔、皮下以及局部注射工艺要求从配制到灭菌,一般应控制在从配制到灭菌应控制在 4 小时内12 小时内达成达成附带剂可加入适合抑菌剂、止痛剂和不得加入任何抑菌剂、止痛剂、增增溶剂溶剂不溶性微除还有规定外,每个供试品容除还有规定外, 1ml 中含 10μm粒器中含 10μ m 以上的微粒不得以上的微粒不得超出 25 粒,含 25超出 6000 粒,含 25μm 以上μ m 以上的微粒不得超出三粒的微粒不得超出 600 粒浸透压等渗等渗、高渗或等张3.影响制剂中药物降解的要素及增添稳固性的方法处方要素: pH 值的影响、广义酸碱催化的影响、溶剂的影响、离子强度的影响、表面活性剂的影响、处方中基质或赋形剂的影响外界要素:温度的影响、光芒的影响、氧的影响、金属离子的影响、湿度和水分的影响、包装资料的影响增添稳固性的方法: 1.改变药物构造(1)、制成难溶性盐(2)、制成复合物(3)、制成前体药物 2.制成固系统剂 3.采纳粉末直接压片或包衣工艺 4.制成微囊、微球或包合物4.片剂制备可能出现的问题及原由剖析1.裂片:处方要素和工艺要素致使片剂内部的压力散布不平均2.松片:片剂硬度不够3.粘冲:干燥不够、4.片重差别超限:物料的流动性差、物猜中细分太多或粒度大小相差悬殊、料斗内的物料时多时少、刮粉器与模孔符合性差5.崩解缓慢:压缩力过大、可溶性成分溶解、片剂的结协力过强、崩解剂的吸水膨胀能力差或对结协力的崩溃能力差6.溶出超限:片剂不崩解、颗粒过硬、药物的溶解度差7.含量不平均:片重差别超限5.片剂的质量检查外观性状;片重差别;硬度和脆碎度;崩解度;溶出度和开释度;含量平均度1.表面活性剂的应用增溶剂;乳化剂;湿润剂;起泡剂和消泡剂;去污剂;消毒剂和杀菌剂剖析题1.维生素 C 注射液的处方剖析处方:维生素 C104g依地酸二钠碳酸氢钠49g注射用水加至 1000ml亚硫酸氢钠2g制备:注射用水80%通CO2+Vc搅拌溶解+粉刺慢慢碳酸氢钠搅拌溶解+(依地酸二钠+亚硫酸氢钠)搅匀pH6.0-6.2+CO2饱和的注射用水至足量滤过,垂熔玻璃漏斗+微孔滤膜+CO2,灌封(CO2或N2 下),灭菌(流通蒸汽100℃ 15min)讲解: 1.临床应用:预防和治疗坏血病作用:减少痛苦、提升稳固性3.抗氧化剂只好改良色彩,对稳固含量无作用。
制剂通则——注射剂注射剂系指药材经提取、纯化后制成的供注入体内的溶液、乳液及供临用前配制或稀释成溶液的粉末或浓溶液的无菌制剂。
注射剂可分注射液、注射用无菌粉末与注射用浓溶液。
注射液系指注射入体内用的无菌溶液型注射液或乳液型注射液。
可用于肌内注射、静脉注射或静脉滴注等。
其中,供静脉滴注用的大体积(除另有规定外,一般不小于100ml)注射液也称静脉输液。
注射用无菌粉末系指供临用前用适宜的无菌溶液配制成溶液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配制后注射,也可用静脉输液配制后静脉滴注。
无菌粉末用冷冻干燥法或喷雾干燥法制得,供直接分装;无菌块状物用冷冻干燥法制得。
注射用浓溶液系指临用前稀释供静脉滴注用的无菌浓溶液。
注射剂在生产与贮藏期间应符合下列有关规定。
一、除另有规定外,药材应按各品种项下规定的方法提取、纯化、制成半成品,以半成品投料配制成品。
二、溶液型注射剂应澄明。
乳液型注射剂应稳定,不得有相分离现象,不得用于椎管注射;静脉用乳液型注射液分散相球粒的粒度90%应在1μm以下,不得有大于5μm的球粒。
静脉输液应尽可能与血液等渗。
三、注射剂所用溶剂必须安全无害,并不得影响疗效和质量,一般分为水性溶剂和非水性溶剂。
(1)水性溶剂最常用的水性溶剂为注射用水,也可用0.9%氯化钠溶液或其他适宜的水溶液。
(2)非水性溶剂常用的非水性溶剂为植物油,主要为供注射用的大豆油,其质量应符合“大豆油(供注射用)”标准;其他还有乙醇、丙二醇、聚乙二醇等的水溶液。
四、配制注射剂时,可根据药物的性质加入适宜的附加剂。
如渗透压调节剂、pH值调节剂、增溶剂、抗氧剂、抑菌剂、乳化剂、助悬剂等。
所用附加剂应不影响药物疗效,避免对检验产生干扰,使用浓度不得引起毒性或过度的刺激。
常用的抗氧剂有亚硫酸钠、亚硫酸氢钠和焦亚硫酸钠,一般浓度为0.1%~0.2%;常用抑菌剂为0.5%苯酚、0.3%甲酚、0.5%三氯叔丁醇等。
多剂量的注射液可加适宜的抑菌剂,抑菌剂的用量应能抑制注射液中微生物的生长,加有抑菌剂的注射液,仍应用适宜的方法灭菌。
第四章药物灭菌制剂和其他制剂与临床应用第一节灭菌制剂一、灭菌制剂和无菌制剂的基本要求1.分类①注射②植入③眼用④局部外用:外伤、烧伤、溃疡等创面⑤手术:冲洗剂、止血海绵剂、骨蜡2.灭菌制剂和无菌制剂的一般质量要求①无菌、无热原②可见异物和不溶性微粒③渗透压(相等或接近)、pH④安全性高(刺激性、毒性反应)⑤稳定性⑥降压物质(抗生素、氨基酸、多肽)二、注射剂1.注射剂的分类①注射液溶液、乳状液、混悬液(不适合中药)皮下、皮内、肌内、静注、静滴(≮100ml)②注射用无菌粉末(粉针):临用前配制③注射用浓溶液:临用前稀释2.注射剂的特点①药效迅速、剂量准确、作用可靠②适用:不宜口服给药的患者/药物③可局部定位④给药不方便,疼痛⑤易发生交叉污染、安全性不及口服制剂⑥生产不便,成本价格高3.注射剂的溶剂①注射用水:注射、滴眼:溶剂、稀释剂、容器清洗②灭菌注射用水:注射用灭菌粉末溶剂、注射剂的稀释剂③注射用油:大豆油、茶油、麻油④其他:乙醇、PG、PEG、甘油4.注射剂的附加剂5.热原污染途径①溶剂带入②原辅料带入③容器、用具带入④制备过程带入——严格按GMP规定操作⑤使用过程带入:注射器具细菌内毒素或热原检查:家兔法(药典法)、鲎试验法除去容器或用具上热原的方法?X:注射剂的优点有 A.药效迅速、剂量准确、作用可靠 B.适用于不宜口服的药物C.适用于不能口服给药的病人D.可迅速终止药物作用E.可以产生定向作用『正确答案』ABCEA:以下有关制药用水说法错误的是 A.纯化水是原水经蒸馏等方法制得的供药用的水 B.纯化水可作配制普通药物制剂的溶剂 C.注射用水是纯化水经蒸馏所得的水 D.注射用水可用于注射用灭菌粉末的溶剂 E.灭菌注射用水可用于注射剂的稀释剂『正确答案』DA.抑菌剂B.等渗调节剂C.抗氧剂D.润湿剂E.助悬剂下列注射剂附加剂的作用是 1.聚山梨酯类 2.羧甲基纤维素 3.硫代硫酸钠 4.葡萄糖 5.苯甲醇『正确答案』D、E、C、B、AA.耐热性B.水溶性C.不挥发性D.过滤性E.不耐强酸、强碱下列除去热原的方法对应的性质分别是 1.蒸馏法制注射用水 2.加入重铬酸钾硫酸清洁液 3.180℃,2小时被破坏『正确答案』C、E、AX:生产注射剂时常加入适量活性炭,其作用为 A.吸附热原 B.能增加主药稳定性 C.脱色 D.脱盐 E.提高澄明度『正确答案』ACE 6.注射剂的临床应用与注意事项①不宜口服给药(患者、药物)、急救、药物没有合适的口服剂型(氨基酸类、胰岛素)②减少注射次数,序贯疗法③减少联合用药④临用配制⑤严格控制剂量、疗程口服-注射-输液7.增加药物溶解度/溶出速度的方法①加入增溶剂(表面活性剂)②加入助溶剂:I2+KI=KI3、咖啡因→苯甲酸钠③使用混合溶剂:潜溶剂(水+乙醇/丙二醇/聚乙二醇/甘油)④制成共晶(阿德福韦酯+糖精形成新晶型)⑤制成盐类:难溶性弱酸、弱碱⑥其他:温度↑,改变pH,微粉化,包合技术维生素C注射液pH值调节剂金属离子螯合剂抗氧剂在维生素C注射液中 A.亚硫酸氢钠 B.二氧化碳 C.碳酸氢钠 D.依地酸二钠 E.注射用水1.适用于偏酸性药液抗氧的是2.用于溶解原辅料的是3.对金属离子有络合作用的是4.与维生素C部分成盐,减轻局部刺激作用的是5.用于除去药液及安瓿空间内氧气的是『正确答案』A、E、D、C、B三、输液1.分类①电解质输液:氯化钠、乳酸钠。
药剂学知到章节测试答案智慧树2023年最新德州学院第一章测试1.关于药物制剂与剂型的说法错误的是()参考答案:注射剂、单剂量滴眼剂中均不得加入抑菌剂2.《中国药典》收载的阿司匹林标准中,记在【性状】项的内容是()参考答案:溶解度3.下列关于药剂学分支学科不包括()。
参考答案:方剂学4.在工作中欲了解化学药物制剂各剂型的基本要求和常规检查的有关内容,需查阅的是()参考答案:《中国药典》四部通则5.对《中国药典》规定的项目与要求的理解,错误的是()参考答案:贮藏条件为“在阴凉处保存”,是指保存温度不超过10℃6.欧洲药典的缩写是()参考答案:EP7.有肝脏首过效应的吸收途径是()。
参考答案:胃粘膜吸收8.《中国药典》最早颁布的时间是()参考答案:1953年9.收载于《中国药典》二部的品种是()参考答案:阿司匹林片10.《中国药典》规定,凡检查溶出度、释放度或分散均匀性的制剂,一般不再检查的项目是()参考答案:崩解时限第二章测试1.药物制剂稳定性变化可分为物理性、化学性和生物性三大分类。
下列稳定性变化中属于物理性变化的是()参考答案:沉降分层2.关于药物制剂稳定性的说法,错误的是()参考答案:药用辅料要求化学性质稳定,所以辅料不影响药物制剂的稳定性3.在水溶液中不稳定,临用时需现配的药物是()参考答案:青霉素钠4.关于稳定性试验的基本要求叙述错误的是()参考答案:加速试验与长期试验适用于原料药与药物制剂,要求用一批供试品进行5.药品的稳定性受到多种因素的影响,下属哪一项为影响药品稳定性的环境因素()参考答案:湿度6.不属于药物稳定性试验方法的是()。
参考答案:随机试验7.关于药物制剂稳定性实验的说法,错误的是()参考答案:加速试验是在温度60℃±2℃和相对湿度75%±5%的条件进行的8.影响药物制剂稳定性的非处方因素是()参考答案:温度9.下面稳定性变化中属于化学稳定性变化的是()参考答案:药物发生水解10.提高药物稳定性的方法有()参考答案:对水溶液不稳定的药物,制成固体制剂;对稳定的有效成分,制成前体药物;对生物制品,制成冻干粉制剂;为防止药物因受环境中的氧气,光线等影响,制成囊或包合物;对遇湿不稳定的药物,制成褒义制剂第三章测试1.不影响乳剂质量的是()参考答案:分层与絮凝2.不属于低分子溶液剂的是()参考答案:布洛芬混悬滴剂3.关于非无菌液体制剂特点的说法,错误的是()参考答案:分散度大,吸收慢4.在配制液体制剂时,为了增加难溶性药物的溶解度,通常需要在溶剂中加入第三种物质,与难溶性药物形成可溶性的分子间络合物,缔合物和复盐等。
注射用无菌粉末俗称粉针,是指药物制成的供临用前用适宜的无菌溶剂配制成澄清溶液或均匀混悬液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配制后注射,也可以用静脉输液配制后静脉滴注。
凡是在水溶液中不稳定的药物,如某些抗生素(青霉素类、头孢菌素类)、一些酶制剂(胰蛋白、辅酶A)及血浆等生物制剂,不能制成水溶性注射剂,更不能在溶液中加热灭菌,均须制成注射用无菌粉末。
近年也将中药注射剂研制成粉针以提高其稳定性,如双黄连粉针、茵栀黄粉针等。
根据生产工艺的不同,注射用无菌粉末可分为两大类:注射用无菌分装制品和注射用冷冻干燥制品。
注射用无菌分装制品注射用无菌分装制品系将采用灭菌溶剂结晶法、喷雾干燥法制得的无菌原料药在无菌条件下直接分装于洁净灭菌的小瓶或安瓿中,密封而成。
常用于抗生素药品,如注射用青霉素钠、注射用头孢呋辛钠等。
(一)制备流程注射用无菌分装制品制备流程,如下图。
(二)基本工艺1、原材料及容器准备无菌原料可在无菌条件下采用结晶法或喷雾干燥法制备,必要时在无菌条件下进行粉碎、过筛等操作,制得符合分装要求的注射用无菌粉末。
安瓿或玻璃瓶与胶塞的处理按注射剂的要求均需进行灭菌处理。
安瓿或玻璃瓶可于180℃干热灭菌1.5小时或于250℃干热灭菌45分钟,胶塞洗净后要用硅油进行处理,再用125℃干热灭菌2.5小时或于121℃湿热,灭菌30分钟灭菌好的空瓶应在净化空气下存放,时间不应超过24小时,具体存放时间应经过验证后再确定。
2、无菌粉末的分装分装必须在A级洁净环境中按无菌操作法进行,分装时多以容积进行定量,可用人工法或机械分装法。
手工分装常采用刮板式分装器,机械分装设备有螺旋式自动分装机、直管式自动分装机和真空吸粉自动分装机等。
分装机宜有局部层流装置。
分装后的小瓶应立即加塞并用铝盖密封。
为了避免铝屑污染产品,轧盖常与分装分开,轧盖则在另一台设备上完成。
若是安瓿,分装后应立即用火焰熔封。
3、灭菌和异物检查对于耐热品种,可选用适宜的灭菌方法进行补充灭菌,以确保安全。