先天性小脑共济失调有哪些症状
- 格式:docx
- 大小:4.29 KB
- 文档页数:2


是以平衡障碍为主,表现为站立或步行时躯体易向患侧倾斜,摇晃不稳,沿直线走时更为明显,改变头位可使症状加重,四肢共济运动多正常。
此外有明显的眩晕、呕吐、眼球震颤。
多见于Meniere病、桥小脑角综合症等
共济失调可分为四种类型:①深感觉障碍性共济失调;②小脑性共济失调;③前庭迷路性共济失调;④大脑型共济失调。
而临床上一般称呼的“共济失调”,多特指小脑性共济失调。
大脑性共济失调通常不如小脑性共济失调症状明显,较少伴发眼球震颤。
大脑性共济失调分为以下三种:
1.额叶性共济失调出现于额叶或额桥小脑束病变时,表现如同小脑性共济失调,如体位性平衡障碍、步态不稳、向后或向一侧倾倒;除有对侧肢体共济失调外,常伴有腱反射亢进、肌张力增高、病理反射阳性,以及精神症状、强握反射和强直性跖反射等额叶损害表现。
2.顶叶性共济失调表现对侧患肢不同程度的共济失调,闭眼时症状明显,深感觉障碍多不重或呈一过性;两侧旁中央小叶后部受损可出现双下肢感觉性共济失调及大小便障碍。
3.颞叶性共济失调较轻,可表现一过性平衡障碍,不易早期发现。
大脑皮质额叶→额桥束
顶叶、枕叶、颞叶→顶枕颞桥束
由大脑皮质额叶起始的纤维组成额桥束;由顶、枕、颞叶起始的纤维组成顶枕颞桥束;这些纤维下行经内囊、大脑脚底的两侧,进入脑桥终止于同侧脑桥核。
脑桥核发出的纤维越过中线,经对侧小脑中脚进入小脑,主要终止于新小脑皮质。
构成了皮质一脑桥一小脑系。

共济失调是什么原因文章目录*一、共济失调是什么原因*二、共济失调如何预防*三、共济失调如何治疗共济失调是什么原因小脑性共济失调1、小脑蚓部损害:常见于小脑蚓部肿瘤,儿童以髓母细胞瘤、星形细胞瘤、室管膜瘤,成人以转移瘤多见。
2、小脑半球损害:常见于肿瘤、转移瘤。
结核瘤或脓肿及血管病等。
3、全小脑共济失调:常见于小脑变性及萎缩等。
深感觉障碍性共济失调1、周围神经病变:常见于多发性神经炎,铅、砷、汞中毒,酒精中毒,代谢性疾病等。
2、后根病变:常见于转移瘤。
3、后索病变:常见于脊髓癣联合变性。
酒精中毒、脊髓压迫症等。
4、丘脑病变:常见于脑血管病。
5、顶叶病变:常见于脑血管病力瘤。
大脑性共济失调常见于大脑额叶、顶叶、颜叶、枕叶、肮脏体部等部位的脑血管病,肿瘤,炎症,外伤,变性性疾病等。
前庭性共济失调常见于急性迷路炎、内耳出血、前庭神经或前庭神经核的急性病变等。
共济失调如何预防遗传性共济失调的预防主要在于遗传咨询,但因这类疾病有多种遗传方式,故遗传咨询目前仍有困难。
因此预防主要是避免近亲结婚;对于有家族史的成员,从儿童起就定期去医院检查,及早发现有无骨骼畸形、眼部症状、心脏病变,以及行走不稳等共济失调症状,以便及早治疗,可能使疾病进展得以延缓或使静止稳定的时期得以延长。
共济失调如何治疗突发共济失调者,检查其有无颅内压增高及脑疝,检测患者意识水平,注意有无瞳孔改变、运动无力或瘫痪、颈部强直或疼痛、呕吐。
注意生命体征变化,尤其是呼吸变化,异常呼吸易导致呼吸暂停。
抬高床头,准备紧急复苏设备、行CT 检查或手术。
注意事项行实验室检查,如血液检查毒物水平、放射性检查,帮助患者适应。
建立康复目标,确保患者安全。
例如感觉性共济失调患者应行动缓慢,利用手杖辅助行走。
向家属了解家庭中有无危害患者健康因素损害,如地面不平整或楼梯栏杆缺失,必要时进一步咨询。
特殊人群儿童可出现急性或慢性共济失调,常见于先天性或获得性疾病。
急性共济失调见于感染、脑肿瘤、腮腺炎及其他疾病。

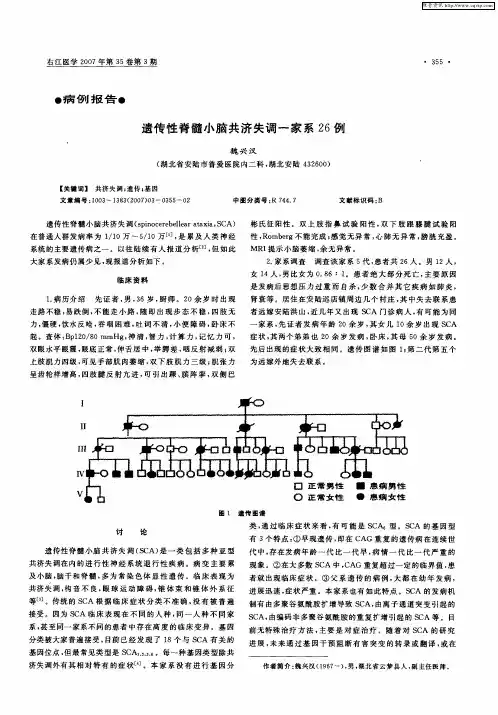
•论著.脊髓小脑性共济失调1型一哈萨克族家系遗传学特征分析承马建华F雷晶*张艳”【摘要】目的探讨脊髓小脑性共济失调1型(spinocerebellar ataxia type1,SCA1)一哈萨克族家系遗传学特征。
方法应用聚合酶链反应、琼脂糖凝胶电泳、T载体连接克隆测序等技术对42例家系成员进行ATXN1基因CAG三核昔酸重复数目测定。
对检测结果进行分析并与表型正常的家系成员和健康人对照。
结果42例家系成员中SCA1基因突变者19例,5例已发病SCA1患者异常等位基因CAG重复数目为41~47次。
家系内表型正常的23例成员SCA1等位基因CAG重复数目为17-30次。
60名健康对照者SCA1等位基因CAG重复数目为12-30次。
在正常等位基因中发现CAT结构的插入。
结论哈萨克族SCA1家系是国内首次报告少数民族SCA1家系,CAG异常扩增数目在不同民族中存在差异。
等位基因中CAT结构的插入更有利于基因的稳定性。
【关键词】脊髓小脑性共济失调1型哈萨克族三核昔酸重复ATXN1基因CAT结构【中图分类号】R744.7【文献标识码】AThe analysis of genetic characteristics of spinocerebellar ataxia type1in a Kazak family.MA Jianhua,LEI Jing,ZHANG Yan.Depatment of Neurologu,,y>the First Affilaied Hospital of Xinjiang Medical University,Urumqi 830054,China.Tel:0991-*******.[Abstract]Objective To explore the genetic characteristics of the spinocerebellar ataxia type1(SC Al)in a Kazakh family.Methods Genetic test(ATXN1gene)of42family members were conducted by polymerase chain reaction,agarose gel electrophoresis and T vector ligand sequencing.Genetic test results were analyzed and comparisonof phenotypes were made between family members and healthy people.Results Among the42family members,19hadthe mutation of SC A l gene,and the number of CAG repeats of the abnonnal allele in5patients with SC A l was41to47, while the number of CAG repeats in the23members with nonnal phenotype in the family was17to30.The number ofthe SC A l allele CAG in60healthy controls were12to30.Insertion of CAT structure found in normal alleles. Conclusion The Kazak SC A l family is the first reported of SC A l in ethnic minority families in China,and the abnormal length of CAG repeats varies among different ethnic groups.The insertion of the CAT structure in the allele tends to increase the stability of the gene.[Key words]Spinocerebeller ataxia type1Kazakh family Triple nucleotide repeats ATXN1gene CAT structure脊髓小脑性共济失调1型(spinocerebellar ataxia type1,SC A l)是一种常染色体显性遗传性神经系统退行性疾病,通常在30〜40岁发病。


共济失调共济失调(ataxia)是指肌力正常的情况下运动的协调障碍。
肢体随意运动的幅度及协调发生紊乱,以及不能维持躯体姿势和平衡。
但不包括肢体轻度瘫痪时出现的协调障碍、眼肌麻痹所致的随意运动偏斜,视觉障碍所致的随意运动困难以及大脑病变引起的失用症。
任何一个简单的运动必须有主动肌、对抗肌、协同肌和固定肌四组肌肉的参与才能完成,并有赖于神经系统的协调和平衡。
共济失调的病因很多,深感觉(深感觉是指感受肌肉、肌腱、关节和韧带等深部结构的本体感觉。
肌肉是处于收缩或舒张状态;肌腱和韧带是否被牵拉以及关节是处于屈曲还是伸直的状态等的感觉。
检查方法①振动觉检查:置振动的128Hz音叉末端于骨突起处(例如内外踝、膝盖、髂前上棘、腕骨或脊椎棘突等处)以试验患者能否察觉。
注意感受的时限,两侧对比。
②位置觉检查:瞩患者闭目,移动患者一肢的大多数关节,塑成一种姿势,瞩患者保持之,然后瞩患者用对侧的一肢模仿。
③运动觉检查:轻移患者的手指和足趾向上及向下,瞩患者说出移动的方向)、前庭系统、小脑和大脑损害都可发生共济失调,根据病变部位不同,共济失调可分为四种类型:①深感觉障碍性共济失调;②前庭迷路性共济失调;③小脑性共济失调;④大脑型共济失调。
而一般称呼的“共济失调”,多特指小脑性共济失调。
几乎100%的Ias(颅内动脉粥样硬化性狭窄)患者有共济失调的表现。
还有原因不明的因素,有的伴有智能不全或痴呆。
神经系统的协调和平衡包括:1.感觉性深感觉向中枢神经系统反映躯体各部位的位置和运动方向。
病因有:①周围神经或神经根病;②脊髓亚急性联合变性(简称亚急性联合变性(subacute combined degeneration,SCD),是由于维生素B12的摄入、吸收、结合、转运或代谢障碍导致体内含量不足而引起的中枢和周围神经系统变性的疾病。
病变主要累及脊髓后索、侧索及周围神经等,临床表现为双下肢深感觉缺失、感觉性共济失调、痉挛性瘫痪及周围性神经病变等,常伴有贫血的临床征象。


脊髓小脑共济失调2型的最新诊疗进展作者:周玲来源:《赤峰学院学报·自然科学版》2019年第05期摘要:脊髓小脑共济失调2型(SCA2)是一种常染色体显性遗传性小脑共济失調,该病因ATXN2基因编码区CAG重复序列异常增多,导致细胞内包涵体中突变蛋白(ataxin-2)的异常积累.临床特点以小脑共济失调为主,在发病早期即有广泛细微的运动和非运动损害.目前尚无有效的治疗手段,以延缓病情发展和对症治疗为主.现就近年来SCA2在临床、病因及诊治等方面的研究进展进行综述.关键词:脊髓小脑共济失调2型;ataxin-2;多聚谷氨酰胺扩展;遗传性共济失调;常染色体显性遗传性小脑共济失调中图分类号:R744.7 ;文献标识码:A ;文章编号:1673-260X(2019)05-0115-05脊髓小脑共济失调2型(SCA2)是常染色体显性遗传共济失调最常见的形式之一.该病由染色体12q23-q24.1上ATXN2基因编码区CAG重复序列扩增,引起蛋白多聚谷氨酰胺(polyQ)的异常积累[1-10].polyQ聚集引起细胞毒性,并丧失其生物学功能,导致小脑、脑干、脊髓和脑皮质中大量神经元的功能障碍和死亡,从而引起以小脑共济失调为主的疾病[11,12].这使得诊断对于临床医生来说具有极大挑战性,特别是在没有家族史的信息时.SCA2目前虽然无法治愈,但一些治疗方案,如物理治疗和神经保护药物对患者仍有益.本文希望通过对SCA2的临床表现、诊断和治疗方法等方面的概述,对临床诊治起到一定的提示作用.1 脊髓小脑共济失调2型的流行病学及遗传学目前已报道大约43种脊髓小脑共济失调类型,与20多个基因突变相关,全球患病率为1:35000人[12].SCA2是古巴[13]、印度[14]、墨西哥[15]和意大利南部[16]最普遍的多发性共济失调,是继SCA3的第二大类脊髓小脑共济失调,占全部病例的15%.SCA2由ATXN2基因第1外显子的编码区CAG重复序列异常扩增引起,正常等位基因N 末端区域为13到31个CAG序列重复,90%的人群为22个序列的重复扩增:[(CAG)8 -CAA-(CAG)4 -CAA-(CAG)8],其中CAA的中断被认为在稳定CAG重复扩增和影响次级RNA结构方面起着关键作用,因而可能与表型变异有关[9,10].携带28-33个重复序列的等位基因被认为是中间扩增,并且可能与肌萎缩侧索硬化症或帕金森综合征相关.ATXN2扩增的等位基因≥35个三倍体重复序列,通常无CAA中断,表现为纯CAG序列重复.SCA2患者CAG 序列重复次数多为37-39个.此外,有文献报道,检测出极端CAG重复扩张(>200),并且与严重的婴儿多系统发育异常相关[17].2 脊髓小脑共济失调2型的临床表现SCA2的临床表现包括广泛的运动和非运动特征,累及小脑、脑干、大脑皮层、基底神经节、脊髓和周围神经等组织.即使SCA2的明确诊断依赖于分子检测,但早期眼扫视减慢、反射异常、姿势或运动的严重震颤、以及早期肌痉挛往往提示SCA2临床特征.2.1 小脑的运动症状SCA2的小脑特征在于慢性进行性共济失调,包括步态共济失调、震颤、小脑构音障碍、辨距不良和轮替障碍[9,18].通常,SCA2首发症状多为步态共济失调(97%),而少数患者为构音障碍.60%-80%的SCA2患者发病年龄与CAG重复个数相关,CAG重复个数越多发病年龄越早[19].Figueroa KP等人发现除了CAG重复个数外,还可能存在隐性修饰等位基因和X染色体连锁修饰等原因[20].2.2 非小脑运动症状2.2.1 眼动紊乱SCA2中动眼障碍最显著的特征是眼球水平运动减慢.眼科临床检查显示超过80%的病例,在古巴超过90%的患者伴有眼水平运动减慢.然而,眼震电图检测发现几乎所有SCA2患者(98%)均有眼扫视减慢,是明确SCA2疾病的特异性眼球运动标志[21].眼扫视速度是SCA2敏感的临床表型,可反应早期脑桥变化,可能是共济失调发作前有用的诊断参数.2.2.2 皮质脊髓束功能障碍SCA2皮质脊髓束受损的主要临床症状包括跖反射亢进和痉挛.Velázquez-PérezL等人在37例无共济障碍的SCA2突变携带者和健康受试者中进行了横断面研究.经临床评估及经颅磁刺激检查发现电刺激经皮质脊髓束向下肢缓慢传导,除可反映多聚谷氨酰胺的神经毒性,还可预测共济失调发作时间[22].在欧洲一项33例SCA2患者的调查研究中,舞蹈病存在于15.15%(N=5),肌张力障碍为27.27%(N=9),帕金森综合征为27.27%(N=9)[23].尚不能完全明确神经系统广泛病变的原因是否与CAG重复扩增有关.2.2.3 外周神经病变周围神经受累在SCA2并不少见.在一项据31例SCA2患者利用电生理检查的前瞻性研究中指出,神经元病是SCA2患者神经受累的主要形式,运动神经元受累较多[24].2.2.4 痛性肌肉痉挛痛性肌肉痉挛在SCA2患者中很常见.88%的患者伴有痉挛,通常影响下肢,其次是腹部和躯干肌.在睡眠期间发生可导致患者频繁觉醒.肌肉痉挛发作年龄与CAG重复大小呈负相关[25].虽然SCA2肌肉痉挛的病理机制尚未完全了解,但根据有关报道,它们是由运动神经元超兴奋性状态的远端部分在微小的轴突损伤后侧支发芽过程引起的[26].2.2.5 睡眠障碍SCA2患者最重要的睡眠障碍包括不宁腿综合征(RLS),周期性腿部运动综合征(PLMS),REM睡眠行为障碍(RBD),失眠和夜间腿部抽筋.多导睡眠监测结果显示睡眠结构明显异常和睡眠效率显著下降[19].2.2.6 认知能力下降SCA2患者显示的认知障碍,涉及语言、视觉空间和执行功能.Olivito G等人利用头颅MRI发现小脑脑皮质退化可能影响认知能力[27].Hernandez-Castillo CR及同事在2016年的报道中指出,SCA2患者小脑白质,内侧丘系和小脑中脚的微结构损伤影响了认知能力[28].2.2.7 精神症状精神症状也是SCA2的常见并发症.对于SCA2患者,最常见的症状包括抑郁症和焦虑状态,而精神病罕见.据报道多达22%的抑郁症状中只有7%符合重度抑郁症的标准.这些特征关联到共济失调严重程度,表明患者的残疾感知对抑郁状态的作用[29].2.2.8 自主神经功能障碍自主功能障碍也很常见,包括姿势性低血压,胃肠道改变,性功能障碍,唾液分泌增加,出汗和流泪.心血管神经生理检查显示大多数患者的迷走神经或交感神经异常.自主神经症状与疾病持续时间,CAG重复个数相关[30].3 脊髓小腦共济失调2型的辅助检查表现在大多数SCA2患者中磁共振成像(MRI)显示严重的橄榄体小脑皮层萎缩(OPCA)模式.TI加权分析SCA2患者显示中脑萎缩显著,包括黑质、基底部、小脑中脚、后脚和延髓楔束核.在幕上室观察白质(WM)或灰质(GM)体积变化没有差异[31].T2加权相显示小脑蚓部、脑桥和岛状、额叶、顶叶和颞皮层的灰质减少,双侧脑海绵状回的灰质损失不明显[32].扩散加权成像(DWI)是一种用于研究疾病早期结构性区域性脑部变化并监测疾病进展的敏感方法,显示底部结构的表观扩散系数增加,包括小脑白质、脑桥、延髓、横向脑桥纤维.最近的DWI研究也显示幕上的区域作为半卵圆中心的水平大脑半球及皮质脊髓束的微观结构的变化[33].脑脊液(CSF)和血液检查有助于SCA2患者的病理生理学研究.在这些患者中,CSF和血清中锌水平显著降低,这似乎与环境缺陷和与扩大的CAG重复相关的未知的生理病理学机制引起的.此外,SCA2患者在CSF中表现出促红细胞生成素的明显减少,表明疾病中内源性神经保护机制的功能障碍.SCA2患者抗氧化平衡的措施表明,晚期氧化蛋白产物和过氧化电位显著增加,以及铁离子在血浆中的还原能力,GSH和总过氧化氢降低.在古巴患者的不同队列中观察到谷胱甘肽S-转移酶(GST)的酶活性增加[19].最近古巴的一项研究中显示SCA2患者的脑脊液中多巴胺及其代谢物显著降低,乙醇胺浓度降低,磷脂代谢改变[26].然而,这些生化改变需要在不同的群体以及具有不同临床阶段的患者中进行确认.4 诊断SCA2的明确诊断必须通过基因检测确定.然而,当基因检测条件有限或患者不同意时,详细的家族史和体格检查可以诊断疑似SCA2患者,如根据特征性的眼球水平扫视严重减缓,眼球震颤频率低等.一般来说,发病年龄和CAG重复个数之间存在明显的负相关,但重复个数不能预测个体的发病年龄或疾病严重程度.必须明确的是发病年龄,严重程度,特定症状和疾病的进展是可变的,不能通过家族史或基因检测来预测.5 治疗目前SCA2的治疗方法仅限于支持治疗,只能改善部分运动和非运动症状,但不能阻止疾病的进展.据报道,多巴胺能和抗胆碱能治疗可能减少震颤,肌张力障碍和运动迟缓,使用麦角乙脲(0.1mg/天,4周)的初步研究表明这种多巴胺能治疗对PLMS和其他睡眠障碍的疗效[34].而镁,奎宁,美西律或高剂量的维生素B可减轻痛性肌肉痉挛.例如,使用高剂量复合维生素B的20名古巴患者中进行的开放性临床试验显示外周神经病变的临床和电生理学标志物显著改善.53%的患者在治疗后肌痉挛明显减轻,认知改变部分恢复.SCA2相关的帕金森综合征患者给予左旋多巴可能有效.针对两个SCA2患者丘脑和丘脑底部给予脑深部电刺激,可改善剧烈震颤.常规治疗难治性肌阵挛可考虑使用吡拉西坦.患有构音障碍的患者可以依靠言语治疗和使用电子设备进行沟通.吞咽困难可能需要胶凝剂以促进摄取液体.经皮内镜胃造瘘术(PEG)在疾病晚期可能是必需的.康复不能阻止疾病进展,但可以改善运动表现.Pérez-Avila等人观察到在6个月内接受了运动训练计划的87名SCA2受试者中,姿势和协调具有显著改善[35].5.1 针对致病的ataxin-2下游利鲁唑是一种用于治疗肌萎缩性侧索硬化症的药物,尚未完全了解其作用机制,但已表明可通过调节谷氨酸神经递质传递和抑制电压门控钠离子通道发挥神经保护作用.2015年,利用利鲁唑进行了一项随机,双盲,安慰剂对照试验,包括16例遗传性共济失调混合样本的SCA2患者,证明利鲁唑耐受性良好,对50%治疗组患者的SARA评分降低,安慰剂组为11%,具有积极的作用.然而,虽然证明了利鲁唑对共济失调严重程度具有疗效,但样本量较小,尚需要大样本和同质的患者队列中确认[36].锌在神经发育,突触可塑性和神经保护的调节方面具有调节作用.2011年,鉴于几乎所有的SCA2患者的锌值都低于世界卫生组织和正常古巴人口所规定的正常范围,表明Zn缺乏可能是导致SCA2发病的一个因素.Velázquez-Pérez及其同事在36名古巴SCA2患者中报道了给予6个月,50mg硫酸锌的双盲和安慰剂对照临床试验,对小脑共济失调,周围神经病变,眼扫视病理学和氧化应激具有改善作用[37].丹曲林,兰尼碱受体抑制剂和细胞内钙离子的稳定剂,可减少浦肯野细胞中谷氨酸诱导的细胞死亡.在SCA2(Q58)鼠模型中证实,丹曲洛林可改善小鼠的运动协调能力并减少浦肯野细胞的损失[38].锂可刺激自噬,清除蛋白质聚集体,降低三磷酸肌醇水平和随后ITPR1的钙流出.但一项随机,安慰剂对照试验显示给予SCA2患者治疗量锂剂后SARA评分及MRI显示脑容积差异无统计学意义[39].5.2 針对突变基因反义寡核苷酸(ASO)治疗可减少polyQ的表达,ASO进入细胞后,可与互补的mRNA 和DNA‐形成RNA双链,识别和清除RNaseH酶.基于这种治疗方法,在转基因小鼠动物模型上给予ASO,发现其部分逆转了运动性能和浦肯野细胞发射率的功效,有希望在未来应用于临床试验[40].此外,ataxin-2异常积累蛋白的减少有助于肌萎缩侧索硬化症和额颞叶痴呆的治疗.TDP‐43的异常包含7%的肌萎缩侧索硬化症和近50%的额颞痴呆,在ataxin-2基因敲除/TDP‐43转基因小鼠,ataxin-2降低减少了TDP‐43聚集和显著提高了小鼠生存率[41].6 结论SCA2为常染色体显性遗传疾病,给患者及其患者的家庭造成终身打击及遗憾,目前的发病机制,神经变性的生理病理学基础和前驱阶段表征仍不完全清楚,尚没有完全根治的治疗方案.此外,临床前治疗尚需与症状现状相结合,导致临床实践还需要解决重要的伦理问题.通过对转基因小鼠模型及SCA2患者的分析结果表明,神经功能障碍先于神经元损害,并且转基因小鼠模型更是提示功能的部分恢复具有年龄依赖性,即使在疾病进展相对较晚的阶段,但只要在神经元损失之前,就有可能具有一些恢复能力.这就强调早期发现,早期干预的重要性,但我们尚需要更多的数据来明确多早给予干预具有改善疾病的效果.参考文献:〔1〕Auburger GW: Spinocerebellar ataxia type 2. Handb Clin Neurol 2012, 103:423-436.〔2〕Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I,Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A et al: Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996, 14(3):269-276.〔3〕Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U: Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012, 124(1):1-21.〔4〕Durr A: Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010, 9(9):885-894.〔5〕Velazquez Perez L, Cruz GS, Santos Falcon N, Enrique Almaguer Mederos L,Escalona Batallan K, Rodriguez Labrada R, Paneque Herrera M, Laffita Mesa JM, Rodriguez Diaz JC, Rodriguez RA et al: Molecular epidemiology of spinocerebellar ataxias in Cuba:insights into SCA2 founder effect in Holguin. Neurosci Lett 2009, 454(2):157-160.〔6〕Faruq M, Scaria V, Singh I, Tyagi S, Srivastava AK, Mukerji M: SCA-LSVD:a repeat-oriented locus-specific variation database for genotype to phenotype correlations in spinocerebellar ataxias. Hum Mutat 2009, 30(7):1037-1042.〔7〕Alonso E, Martinez-Ruano L, De Biase I, Mader C, Ochoa A, Yescas P,Gutierrez R, White M, Ruano L, Fragoso-Benitez M et al: Distinct distribution of autosomal dominant spinocerebellar ataxia in the Mexican population. Mov Disord 2007, 22(7):1050-1053.〔8〕Brusco A, Gellera C, Cagnoli C, Saluto A, Castucci A, Michielotto C, Fetoni V, Mariotti C, Migone N, Di Donato S et al: Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch Neurol 2004, 61(5):727-733.〔9〕Di Fabio R, Santorelli F, Bertini E, Balestri M, Cursi L, Tessa A, Pierelli F,Casali C: Infantile childhood onset of spinocerebellar ataxia type 2. Cerebellum 2012, 11(2):526-530.〔10〕Maas RP, van Gaalen J, Klockgether T, van de Warrenburg BP: The preclinical stage of spinocerebellar ataxias. Neurology 2015, 85(1):96-103.〔11〕Velazquez-Perez L, Rodriguez-Labrada R, Garcia-Rodriguez JC, Almaguer-Mederos LE, Cruz-Marino T, Laffita-Mesa JM: A comprehensive review of spinocerebellar ataxia type 2 in Cuba. Cerebellum 2011, 10(2):184-198.〔12〕Figueroa KP, Coon H, Santos N, Velazquez L, Mederos LA, Pulst SM:Genetic analysis of age at onset variation in spinocerebellar ataxia type 2. Neurol Genet 2017, 3(3):e155.〔13〕Rodriguez-Labrada R, Velazquez-Perez L, Auburger G, Ziemann U, Canales-Ochoa N, Medrano-Montero J, Vazquez-Mojena Y, Gonzalez-Zaldivar Y: Spinocerebellar ataxia type 2: Measures of saccade changes improve power for clinical trials. Mov Disord 2016,31(4):570-578.〔14〕Velazquez-Perez L, Rodriguez-Labrada R, Torres-Vega R, Medrano Montero J,Vazquez-Mojena Y, Auburger G, Ziemann U: Abnormal corticospinal tract function and motor cortex excitability in non-ataxic SCA2 mutation carriers: A TMS study. Clin Neurophysiol 2016,127(8):2713-2719.〔15〕Pedroso JL, Braga-Neto P, Escorcio-Bezerra ML, Abrahao A, de Albuquerque MV, Filho FM, de Souza PV, de Rezende Pinto WB, Borges FR, Jr., Saraiva-Pereira ML et al: Non-motor and Extracerebellar Features in Spinocerebellar Ataxia Type 2. Cerebellum 2017,16(1):34-39.〔16〕Bezerra ML, Pedroso JL, Braga-Neto P, Abrahao A, de Albuquerque MV,Borges FR, Jr., Saraiva-Pereira ;ML, Jardim LB, de Oliveira Braga NI, Manzano GM et al:Pattern of Peripheral Nerve Involvement in Spinocerebellar Ataxia Type 2: a Neurophysiological Assessment. Cerebellum 2016, 15(6):767-773.〔17〕Velazquez-Perez L, Rodriguez-Labrada R, Canales-Ochoa N, Montero JM,Sanchez-Cruz G, Aguilera-Rodriguez R, Almaguer-Mederos LE, Laffita-Mesa JM:Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol 2014, 13(5):482-489.〔18〕Velazquez-Perez L, Tunnerhoff J, Rodriguez-Labrada R, Torres-Vega R,Belardinelli P, Medrano-Montero J, Pena-Acosta A, Canales-Ochoa N, Vazquez-Mojena Y,Gonzalez-Zaldivar Y et al: Corticomuscular Coherence: a Novel Tool to Assess the Pyramidal Tract Dysfunction in Spinocerebellar Ataxia Type 2. Cerebellum 2017, 16(2):602-606.〔19〕Olivito G, Lupo M, Iacobacci C, Clausi S, Romano S, Masciullo M, Molinari M, Cercignani M, Bozzali M, Leggio M: Microstructural MRI basis of the cognitive functions in patients with Spinocerebellar ataxia type 2. Neuroscience 2017.〔20〕Hernandez-Castillo CR, Vaca-Palomares I, Galvez V, Campos-Romo A, Diaz R,Fernandez-Ruiz J: Cognitive Deficits Correlate with White Matter Deterioration in Spinocerebellar Ataxia Type 2. J Int Neuropsychol Soc 2016, 22(4):486-491.〔21〕Lo RY, Figueroa KP, Pulst SM, Perlman S, Wilmot G, Gomez C,Schmahmann J, Paulson H, Shakkottai VG, Ying S et al: Depression and clinical progressionin spinocerebellar ataxias. Parkinsonism Relat Disord 2016, 22:87-92.〔22〕Montes-Brown J, Sanchez-Cruz G, Garcia AM, Baez ME, Velazquez-Perez L:Heart rate variability in type 2 spinocerebellar ataxia. Acta Neurol Scand 2010, 122(5):329-335.〔23〕Mascalchi M, Diciotti S, Giannelli M, Ginestroni A, Soricelli A, Nicolai E,Aiello M, Tessa C, Galli L, Dotti MT et al: Progression of brain atrophy in spinocerebellar ataxia type 2: a longitudinal tensor-based morphometry study. PLoS One 2014, 9(2):e89410.〔24〕Mercadillo RE, Galvez V, Diaz R, Hernandez-Castillo CR, Campos-Romo A,Boll MC, Pasaye EH, Fernandez-Ruiz J: Parahippocampal gray matter alterations in Spinocerebellar Ataxia Type 2 identified by voxel based morphometry. J Neurol Sci 2014, 347(1-2):50-58.〔25〕Della Nave R, Ginestroni A, Tessa C, Cosottini M, Giannelli M, Salvatore E,Sartucci F, De Michele G, Dotti MT, Piacentini S et al: Brain structural damage in spinocerebellar ataxia type 2. A voxel-based morphometry study. Mov Disord 2008, 23(6):899-903.〔26〕Velazquez-Perez L, Rodriguez-Labrada R, Alvarez-Gonzalez L, Aguilera-Rodriguez R, Alvarez Sanchez M, Canales-Ochoa N, Galicia Polo L, Haro-Valencia R,Medrano-Montero J, Vazquez-Mojena Y et al: Lisuride reduces involuntary periodic leg movements in spinocerebellar ataxia type 2 patients. Cerebellum 2012, 11(4):1051-1056.〔27〕Perez-Avila I, Fernandez-Vieitez JA, Martinez-Gongora E, Ochoa-Mastrapa R,Velazquez-Manresa MG: [Effects of a physical training program on quantitative neurological indices in mild stage type 2 spinocerebelar ataxia patients]. Rev Neurol 2004, 39(10):907-910.〔28〕Romano S, Coarelli G, Marcotulli C, Leonardi L, Piccolo F, Spadaro M,Frontali M, Ferraldeschi M, Vulpiani MC, Ponzelli F et al: Riluzole in patients with hereditarycerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015, 14(10):985-991.〔29〕Velazquez-Perez L, Rodriguez-Chanfrau J, Garcia-Rodriguez JC, Sanchez-Cruz G, Aguilera-Rodriguez R, Rodriguez-Labrada R, Rodriguez-Diaz JC, Canales-Ochoa N,Gotay DA, Almaguer Mederos LE et al: Oral zinc sulphate supplementation for six months in SCA2 patients: a randomized, double-blind, placebo-controlled trial. Neurochem Res 2011, 36(10):1793-1800.〔30〕Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM,Bezprozvanny I: Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci 2009, 29(29):9148-9162.〔31〕Sacca F, Puorro G, Brunetti A, Capasso G, Cervo A, Cocozza S, de Leva M,Marsili A, Pane C, Quarantelli M et al: A randomized controlled pilot trial of lithium in spinocerebellar ataxia type 2. J Neurol 2015, 262(1):149-153.〔32〕Pulst SM: Degenerative ataxias, from genes to therapies: The 2015 Cotzias Lecture. Neurology 2016, 86(24):2284-2290.〔33〕Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G et al: Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544(7650):367-371.。


多系统萎缩的症状有哪些?常见症状:小脑性共济失调、皮质功能减退、共济失调、植物神经功能障碍1.纹状体黑质变性(1)中年起病,男性多见。
主要表现进行性肌强直、运动迟缓和步态障碍,病情发展到后期可导致自主神经损害、锥体束损害及(或)小脑损害。
开始多为一侧肢体僵硬、少动,病情逐渐发展至对侧,导致动作缓慢、步态前冲、转变姿势困难、上肢固定、少摆动、讲话慢及语音低沉等,但震颤很轻或缺如,可有位置性震颤,表现酷似Parkinson病,但大部分患者用左旋多巴治疗无效。
(2)随着病情发展常出现步态不稳、共济失调等小脑体征,以及尿频、尿急、尿失禁、尿潴留、发汗障碍、体位性晕厥和性功能不全等自主神经功能障碍。
少数可有锥体束征、双眼向上凝视困难、肌阵挛、呼吸和睡眠障碍等。
(3)CT检查可见双侧壳核低密度灶。
MCI显示壳核、苍白球T2低信号,提示铁沉着。
2.橄榄脑桥小脑萎缩以明显的桥脑及小脑萎缩为病理特点,多为散发病例,部分病例呈家族性发病,为常染色体显性遗传,称为家族性OPCA。
(1)一般未成年起病,男女均可受累,缓慢进展。
主要表现小脑性共济失调和脑干功能受损,可有自主神经损害、Parkinson综合征、锥体束征等。
如明显步态不稳、基底加宽、眼球震颤和意向性震颤,后期出现肌张力增高、腱反射亢进、Babinski征等锥体束征,波及延髓肌群出现吞咽困难、呛咳、构音障碍和舌肌束颤。
可见强直、震颤、运动缓慢等锥体外系症状,罕有软腭阵挛;少数可有眼肌瘫痪,表现眼球向上或向下凝视麻痹,慢眼球运动可能是OPCA特征性表现。
可有性功能不良、尿失禁、晕厥,以及视神经萎缩等。
(2)病程中晚期MRI可清晰显示小脑、脑干萎缩,第四脑室和脑池扩张。
3.Shy-Drager综合征也称特发性直立性低血压,与前交感神经元变性有关,特征性临床表现是进行性自主神经功能异常,常合并Parkinson病、小脑性共济失调、锥体束征或下运动神经元体征如肌萎缩等。
小脑萎缩的知识问答一、基础知识篇1. 什么是小脑萎缩?小脑萎缩是一种影像学表现,指小脑的容积减小,脑沟增宽。
它可以是由多种原因引起的,包括遗传性、变性性、缺血缺氧性、药物中毒、炎症性等因素。
2. 小脑的主要功能是什么?小脑萎缩会对这些功能产生什么影响?小脑主要负责协调随意运动、调节肌肉张力、维持身体平衡。
当小脑萎缩时,患者会出现平衡失调,比如站立不稳、走路摇晃,像喝醉酒一样(共济失调);动作不协调,如手部动作笨拙,不能完成精细动作,像写字、扣纽扣等变得困难;还可能出现言语障碍,表现为吟诗样语言或爆发性语言。
3. 小脑萎缩是如何分类的?按病因分类:遗传性小脑萎缩,如脊髓小脑性共济失调(SCA),这是一组常染色体显性遗传病,不同的基因缺陷会导致不同类型的SCA。
获得性小脑萎缩,包括脑血管病引起的缺血性小脑萎缩、酒精中毒导致的酒精性小脑萎缩、药物中毒引起的小脑萎缩(如某些抗肿瘤药、抗癫痫药)、炎症性疾病(如急性小脑炎后遗留的小脑萎缩)等。
按萎缩范围分类:局限性小脑萎缩,仅小脑局部区域出现萎缩。
全小脑萎缩,整个小脑都受到影响。
二、病因篇1. 常见的引起小脑萎缩的遗传性因素有哪些?脊髓小脑性共济失调(SCA)是最常见的遗传性小脑萎缩病因。
目前已经发现了多种致病基因,如SCA1、SCA2、SCA3等。
这些基因的突变会导致神经元变性死亡,从而引起小脑萎缩。
另外,Friedreich共济失调也是一种遗传性疾病,常伴有脊髓后索和小脑的萎缩。
2. 哪些非遗传性因素可能导致小脑萎缩?脑血管因素:小脑梗死或出血后,局部脑组织缺血缺氧坏死,随着时间推移可能导致萎缩。
例如,小脑后下动脉梗死,会影响小脑的血液供应,长期可能引起萎缩。
中毒因素:长期大量饮酒,酒精及其代谢产物会对小脑的神经细胞产生毒性作用,引起酒精性小脑萎缩。
某些药物如顺铂(一种化疗药物)、苯妥英钠(抗癫痫药)等,在长期或过量使用时,也可能损害小脑细胞导致萎缩。
脊髓小脑性共济失调的研究进展崔海燕,康龙丽*,李旭光,朱敏霞,戎浩(西藏民族学院医学院,高原环境与疾病相关基因研究省级重点实验室,生命科学基础实验室,咸阳712082)摘要:脊髓小脑性共济失调是一类遗传性神经系统变性疾病,目前已知的脊髓小脑性共济失调亚型有30余种(DRPLA,SCA1-8,10-23, 25-31)。
基因表达异常,谷氨酸及钙离子依赖性细胞信号转导异常是引起小脑功能紊乱的重要原因。
关键词:脊髓小脑性共济失调;神经变性疾病中图分类号: R744. 7文献标识码: AThe study progress of spinocerebellar ataxiaCUI Hai-yan, KANG Long-li, LI Xu-guang, ZHU Min-xia, RONG Hao (Medical Department of Tibet Nationality College,Plateau Environment and Genes Related to Diseases Key Laboratory of Tibet,Life Science Base laboratory, Xianyang 712082, China)Abstract: Spinocerebellar ataxias is a group of hereditary neurodegenerative disorders. To date, more than 30 kinds of subtypes of genetic spinocerebellar have been identified (DRPLA,SCA1-8,10-23,25-31). Both gene expression and glutamate-dependent and calcium-dependent neuronal signaling as important pathways leading to cerebellar dysfunction.Key words:Spinocerebellar ataxias ; Neurodegenerative disorders脊髓小脑性共济失调(spinocerebellar ataxias ,SCAs)是一组以共济失调为特征的常染色体显性遗传性神经系统变性疾病,病变除了累及脊髓、小脑外,其它组织如脊神经、脑神经、交感神经、基底神经节、丘脑、丘脑下部、大脑皮层均可受累,因此,许多患者除了共济失调外还有其它神经系统症状。
如对您有帮助,可购买打赏,谢谢
先天性小脑共济失调有哪些症状
导语:先天性小脑共济失调是一种先天性疾病,相信很多朋友对先天性小脑共济失调这种疾病不怎么了解,那么先天性小脑共济失调有哪些症状呢?接下来
先天性小脑共济失调是一种先天性疾病,相信很多朋友对先天性小脑共济失调这种疾病不怎么了解,那么先天性小脑共济失调有哪些症状呢?接下来,本文就为大家介绍先天性小脑共济失调的相关症状,想要了解先天性小脑共济失调有哪些症状的朋友可以来看一看哦!下面请大家看详细的介绍。
先天性小脑共济失调有哪些症状?
先天性小脑共济失调多见于1~4岁小儿,偶见于10岁以上。
主要表现为共济失调,常伴有四肢震颤、眼震、肌张力减低、腱反射减弱等。
1、前驱感染史
大部分病例在共济失调发生以前1~3周有前驱感染史,如发热,呼吸道或消化道症状。
约50%病例有发疹性病毒感染史。
有少数病例先有共济失调,10~20天后出现发疹性疾病。
2、起病特点
本病起病急,多以躯干和四肢共济失调开始,很快发展到症状的高峰,表现为站立不稳,步态蹒跚,易于跌倒。
严重者不能站立,完全不能行走,甚至不能独坐、不能竖头。
3、体检
肢体共济失调还可表现为指鼻试验和跟膝胫试验不稳、轮替试验不能、辨距不良及意向性震颤等。
常伴构音障碍。
肌张力及腱反射的减低常不典型、肌力正常。
感觉检查正常,脑神经多不受累。
少数病儿
预防疾病常识分享,对您有帮助可购买打赏。