逆转录病毒载体pOZ—KLF5的构建及鉴定
- 格式:doc
- 大小:34.00 KB
- 文档页数:4

293T逆转录病毒制备(慢病毒包装)1.转染前一天,10%FBS DMEM不含双抗培养基种3*10^6细胞至10cm dish,培养24h(3盘 NC #3 #4)2. a.用OptiMEM 470ul稀释Fugene9 30ul(NC/Sh#3/Sh#4),充分吹打混匀,RT5minb.Target plasmid:3.75ug (NC/Sh#3/Sh#4)Gag-pol:3.75ugVSVG:2.5ug(8/5ug pCDH 6/3.33ug psPAX2 2/1.67ug PMD)c. 将上述准备的质粒混合液滴加到optimem与Fugene9的混合液中,RT静置20min;转染混合物逐滴加至细胞中3. 转染24h之后,弃上清液,更换新鲜培养基,置37°细胞培养箱中继续培养病毒液的收集1.转染48h 后(此时293T长满),收集上清液于15ml 离心管中,3000rpm离心20min2.将上清液用0.45um滤头过滤,再向10cm dish 加10ml培液3.24h之后,再次收集上清液,0.45um滤头过滤,随后保存于-80°冰箱中。
慢病毒感染目的细胞1.需要提前一天铺板至8个10cm dish中(细胞接种个数是1*10^6),以保证感染时的密度为30-40%;(Control,NC,#3,#4 PLC和7405各四盘)2.贴壁后弃上清液,配置25ml+40ul (10mg/ml)polybrene混匀(24ml)培养基DMEM (10%FBS不含双抗) , 6个dish加3ml混匀液,然后加入3ml病毒2盘Control组只加3ml混匀液+3ml DMEM(10%FBS不含双抗)10cm dish置于细胞培养箱中再培养48h3.48h后,弃上清,更换各自新鲜培养液8ml,并加入puromycin筛选PLC/PRE/5 0.6ug/ml(4.8ul 1mg/ml puromycin)Bel-7405 0.5ug/ml(4ul 1mg/ml puromycin)4.筛选2-3天之后,观察dish中细胞生长的状况,判断筛选是否成功(比较Control和感染组生存情况)5.若筛选成功,继续加入相应浓度抗生素筛选(注意杀菌)6.WB验证。

首次应用反转录病毒仅感染分裂细胞特性构建hTERT驱动野生型p53基因反转录病毒载体质粒余新;洪葵;邓俊晖;刘天德;雷钧;张吉翔;邵江华【期刊名称】《中国组织工程研究》【年(卷),期】2008(012)003【摘要】目的:选择只感染分裂细胞的鼠源性反转录病毒载体pLHCX作为载体,构建由hTERT启动子驱动目的基因表达的载体,实现靶向性表达.方法:实验于2005-09/2006-05在江西省分子医学重点实验室完成.①实验材料:含319 bp hTERT核心启动子的phTERT-luc质粒由军事科学院郑晓飞博士惠赠,反转录病毒载体质粒pLHCX由浙江大学朱永良教授惠赠,含1.8 kb wtp53基因的质粒pCMV-Neo-BamH-wtp53由第二军医大学朱明华教授惠赠,增强型绿色荧光蛋白报告质粒pEGFP-N2和pEGFP-C1由本室保存,对照反转录病毒质粒pLHCX-CMV-EGFP 前期构建完成;端粒酶阳性人大肠癌细胞SW620、HT-29,人宫颈癌细胞Hela,人肺癌细胞A549和端粒酶阴性人胚肺成纤维细胞MRC-5由本室保存.②实验方法:利用定向克隆的方法分别构建由hTERT启动子驱动EGFP的质粒pLHCX-hTERT-EGFP和驱动wtp53表达的质粒pLHCX-hTERT-wtp53.以质粒pLHCX-hTERT-EGFP和课题组前期构建的质粒pLHCX-CMV-EGFP转染端粒酶阳性的人大肠癌细胞株SW620和HT-29、人宫颈癌细胞株Hela和人肺癌细胞株A549以及端粒酶阴性的正常人肺胚成纤维细胞MRC-5.③实验评估:通过流式细胞仪测定分析转染效率,证实pLHCX-hTERT-EGFP在不同细胞中的表达特异性.运用RT-PCR和Western Blot分别检测p53 mRNA和p53蛋白在p53突变的大肠癌细胞株SW620中表达.利用MTT检测质粒pLHCX-hTERT-wtp53对不同细胞的增殖抑制情况和流式细胞术检测不同细胞的凋亡情况.结果:①通过酶切证实重组质粒pLHCX-hTERT-EGFP和pLHCX-hTERT-wtp53构建成功,并通过将pLHCX-hTERT-EGFP转染SW620细胞观察到绿色荧光蛋白的表达,测序证实构建质粒中包含完整的wtp53片段.②流式细胞术检测结果证实质粒pLHCX-hTERT-EGFP处理后端粒酶阳性的SW620、Hela和A549内增强型绿色荧光蛋白表达强度明显强于端粒酶阴性的MRC-5(P < 0.05),而pLHCX-CMV-EGFP在端粒阳性和阴性的上述细胞中表达无明显差异(P > 0.05).③RT-PCR和Western Blot分别证实了p53mRNA和p53蛋白表达.④MTT检测到重组质粒对端粒酶阳性的大肠癌细胞有明显的增殖抑制作用,而对端粒酶阴性的MRC-5细胞生长无明显影响(P <0.05).⑤流式细胞检测观察到重组质粒引起端粒酶阳性的大肠癌细胞凋亡要明显强于端粒酶阴性的MRC-5细胞(P < 0.05).结论:构建的反转录病毒载体质粒pLHCX-hTERT-EGFP在端粒酶阳性的肿瘤细胞中特异性表达;重组反转录病毒载体质粒pLHCX-hTERT-wtp53转染后对端粒酶阳性的大肠癌细胞有特异性的影响作用.【总页数】5页(P493-497)【作者】余新;洪葵;邓俊晖;刘天德;雷钧;张吉翔;邵江华【作者单位】南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院江西省分子医学重点实验室,江西省南昌市,330006;南昌大学第二附属医院普外科,江西省南昌市,330006【正文语种】中文【中图分类】R394.2【相关文献】1.卵巢癌的p53基因治疗实验研究——人野生型p53基因真核细胞表达质粒的构建 [J], 关婷;崔满华;李守柔2.小鼠microRNA-31反转录病毒载体重组质粒的构建及鉴定 [J], 张璐玢;谢梦晓;陈云鹏;徐晓昱;张彩云;冯晖晖;刘霞;周小明;夏圣3.抑制人喉癌细胞生长的p53基因反转录病毒重组体的构建 [J], 陈晓巍;曹克利;刘德培4.反转录病毒介导的野生型p53基因诱发人喉癌细胞凋亡的… [J], 陈晓巍;王直中5.由反转录病毒载体转移的马立克病毒糖蛋白D基因在感染细胞中的表达 [J], 段玉友;崔治中;秦爱建;殷震;杨冠珍;吴祥甫因版权原因,仅展示原文概要,查看原文内容请购买。

TIEG特异的siRNA慢病毒载体的构建及鉴定叶礼红;舒晓春;邬伟民;鲁红云;孟晓军;孙辽【期刊名称】《中国现代医学杂志》【年(卷),期】2009(19)17【摘要】目的构建TIEG基因siRNA慢病毒载体.方法针对已经筛选确定的TIEG 基因siRNA有效靶序列.合成靶序列的Oligo DNA退火形成双链DNA与经BamH Ⅰ和EcoRl酶切后的PshRNA-copGFP-lendvector慢病毒载体[含H1启动子和绿色荧光蛋白(GFP)]连接产生psiRNA-TIEG慢病毒载体,PCR筛选阳性克隆.测序鉴定,用psiRNA-TIEG和病毒包装系统共转染包装细胞293T细胞,包装产生慢病毒,以293T细胞GFP蛋白的表达水平测定病毒滴度;用包装病毒颗粒注射大鼠,通过RealTime-PCK检测大鼠肾组织TIEG mRNA的表达水平.结果 PCR和测序证实,成功构建TIEG siRNA慢病毒栽体psiRNA-TIEG,包装慢病毒,浓缩病毒悬液的滴度为1×10<'5>ifu/μL;实验组大鼠TIEGmRNA的表达水平明显低于对照组(50.7%).结论成功构建大鼠TIEG基因siRNA慢病毒栽体psiRNA-TIEG.【总页数】5页(P2568-2572)【作者】叶礼红;舒晓春;邬伟民;鲁红云;孟晓军;孙辽【作者单位】中山大学附属第五医院内分泌科,广东,珠海,519000;中山大学附属第五医院内分泌科,广东,珠海,519000;中山大学附属第五医院内分泌科,广东,珠海,519000;中山大学附属第五医院内分泌科,广东,珠海,519000;中山大学附属第五医院内分泌科,广东,珠海,519000;中山大学附属第五医院内分泌科,广东,珠海,519000【正文语种】中文【中图分类】Q782【相关文献】1.表达HBsAg特异性siRNA的重组腺相关病毒载体的构建 [J], 胡斌;杨燕;刘嘉;马智勇;黄红平;余源;刘慎沛;杨东亮2.C-erbB-2特异性siRNA逆转录病毒载体的构建与鉴定 [J], 陈敏;冯定庆;祝怀平;凌斌;周颖;朱园园;高婷;程志祥;沈国栋;赵卫东3.SMO特异性siRNA慢病毒载体的筛选构建 [J], 杨波;陈卫华;温机灵;华咏;王跃闽4.ABCE1特异性siRNA慢病毒载体的构建与鉴定 [J], 任翼;刘永煜;郭微;田大力5.大鼠NgR特异性siRNA慢病毒载体的构建与鉴定 [J], 林如英;王玮因版权原因,仅展示原文概要,查看原文内容请购买。

轮状病毒NSP5真核表达载体的构建、鉴定及功能研究陈林林; 周艳; 林晓晨; 杜静; 刘洋; 汪艺; 李鸿钧【期刊名称】《《生物学杂志》》【年(卷),期】2019(036)006【总页数】5页(P12-16)【关键词】轮状病毒; NSP5; 真核表达载体; 病毒复制【作者】陈林林; 周艳; 林晓晨; 杜静; 刘洋; 汪艺; 李鸿钧【作者单位】中国医学科学院北京协和医学院医学生物学研究所云南省重大传染病疫苗研发重点实验室昆明650118【正文语种】中文【中图分类】Q78; R373.2轮状病毒(Rotavirus,RV)是导致婴幼儿急性腹泻的重要病原体之一,于1973年在严重腹泻的婴幼儿十二指肠切片及粪便样本中首次被发现,且因其形似车轮,故被命名为轮状病毒[1-3]。
RV的流行和感染多发生在气温较低的秋冬季节,主要通过粪口途径传播。
感染后常导致婴幼儿患者产生腹泻、呕吐、发烧等症状,并伴随严重脱水、体内电解质代谢紊乱等并发症,在全球范围尤其是在南亚、中非等广大发展中国家和地区,严重威胁着当地婴幼儿的健康[4-5]。
RV属呼肠孤病毒科(Reoviridae)、轮状病毒属。
RV无包膜结构,其基因组由11段不连续的双链RNA组成,分别编码着6种结构蛋白(VP1、VP2、VP3、VP4、VP6、VP7)和6种非结构蛋白(NSP1、NSP2、NSP3、NSP4、NSP5、NSP6),在RV宿主特异性、基因组的复制和翻译、宿主胞质内病毒池(viroplasms)的形成及病毒增殖和释放等环节中发挥重要作用[6-8]。
此外,依据结构蛋白VP6的抗原特异性,可将RV分成8组,且因结构蛋白VP4和VP7的差异,RV又被细分成37个P型与27个G型[9-13]。
非结构蛋白NSP5为重要的胞质磷蛋白,具有高度保守的氨基酸序列[14-15]。
在RV感染细胞后,NSP5在胞质内表达并参与形成病毒池结构,调控RV的复制和包装,但具体机制还不完全清楚[16]。
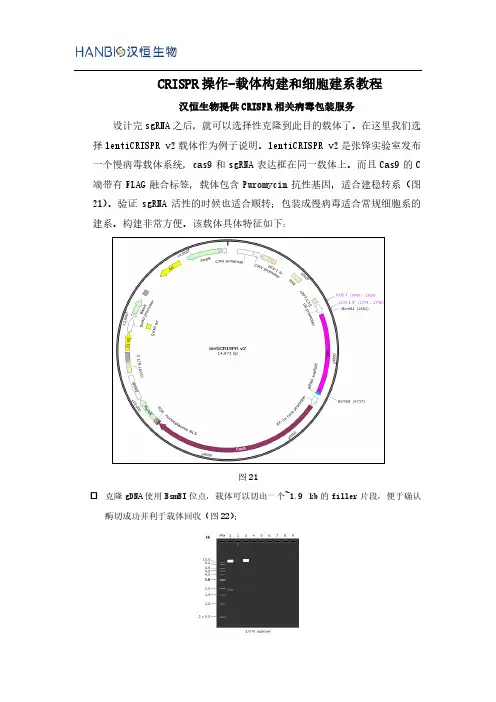
CRISPR操作-载体构建和细胞建系教程汉恒生物提供CRISPR相关病毒包装服务设计完sgRNA之后,就可以选择性克隆到此目的载体了。
在这里我们选择lentiCRISPR v2载体作为例子说明。
lentiCRISPR v2是张锋实验室发布一个慢病毒载体系统,cas9和sgRNA表达框在同一载体上。
而且Cas9的C 端带有FLAG融合标签,载体包含Puromycin抗性基因,适合建稳转系(图21)。
验证sgRNA活性的时候也适合顺转;包装成慢病毒适合常规细胞系的建系。
构建非常方便。
该载体具体特征如下:图21克隆gDNA使用BsmBI位点,载体可以切出一个~1.9 kb的filler片段,便于确认酶切成功并利于载体回收(图22);图22虚拟酶切示意图。
Lane1是BsmBI酶切的虚拟结果;lane3是对照。
☐BsmBI的位点是CGTCTC(1/5)^,别序列和切割序列分离,酶切之后不需要CIP脱磷也不会自连!☐Cas9的COOH 端带有FLAG tag,可以WB或者Immunostaining;☐EFS promoter被优化的小而强,极大提高了慢病毒包装的效率;☐载体带有Puromycin抗性,可以富集转染/感染成功的细胞。
具体克隆步骤如下:1.克隆到lentiCRISPR v2载体的sgRNA格式如下(图23):图23 lentiCRISPR v2载体兼容的sgRNA格式。
举个实例说明(图24):我们锁定NGG之后,只需跟前上游20 bp的序列设计如图所示的oligo即可。
图24 lentiCRISPR v2载体兼容的sgRNA格式实例。
最上面是对应的基因组双链,我们确定NGG之后只需要在前面找到20bp序列即可。
2.然后合成如下两条引物(oligo)即可(注意方向):Oligo1: CACCG/CAGTCTGATCAGTTTTCCTTOligo2: AAACAAGGAAAACTGATCAGACTGC CGTCAGACTAGTCAAAAGGAACAAA3.然后按照如下步骤退火(图25)。

USP22 ShRNA慢病毒载体的构建及鉴定目的构建并鉴定USP22基因ShRNA慢病毒载体,为进一步研究USP22基因在鼻咽癌中的作用机制奠定基础。
方法针对USP22基因的编码序列设计并合成2条特异性干扰序列,序列两端含有限制性内切酶位点HpaⅠ和XhoⅠ。
寡核苷酸链退火生成寡核苷酸双链,5′端磷酸化后将含有酶切位点的寡核苷酸双链克隆到pLL3.7慢病毒表达载体。
连接产物经转化、培养,提取其质粒,提取出来的质粒经HpaⅠ和XhoⅠ酶切电泳鉴定,鉴定正确的质粒进行测序。
构建成功的慢病毒表达载体pLL-USP22-shRNA與包装载体质粒混匀共转染于293T细胞。
通过荧光显微镜下观察绿色荧光蛋白(GFP)情况,对病毒滴度和感染效率进行检测。
结果成功构建慢病毒表达载体pLL-USP22-shRNA。
与包装载体质粒共转染293T细胞后测定慢病毒滴度为4×107 TU/ml。
结论本实验应用相关技术成功构建USP22 ShRNA慢病毒载体,为进一步研究USP22基因的生物学功能奠定了基础。
标签:USP22;慢病毒载体;构建;鉴定肿瘤细胞中基因表达具有组织特异性,USP22泛素水解酶属去泛素化酶DUB基因家族成员,其普遍表达表明其功能的保守性,因此,USP22被归类为肿瘤干细胞的标记基因而引起高度关注[1]。
国内外学者研究发现,USP22基因过表达与结直肠癌[2]、肺癌[3]、胃癌[4]、食管癌[5]、乳腺癌[6]等恶性肿瘤的浸润、转移和预后差高度相关。
沉默USP22基因表达,能显著抑制膀胱癌[7]、结直肠癌[8]细胞增殖,由此推测USP22基因可能成为肿瘤治疗的一个新靶点。
本研究通过基因工程技术构建USP22 ShRNA慢病毒载体,为进一步研究USP22基因在人鼻咽癌细胞中的作用机制提供实验基础。
1 材料与方法1.1 实验材料、试剂及仪器pLL3.7慢病毒表达载体及包装载体质粒购自广州永诺生物科技有限公司。

小鼠survivin基因慢病毒表达载体的构建及鉴定杨平;苟欣;周青松;何卫阳;余昆【期刊名称】《中国组织工程研究》【年(卷),期】2011(015)049【摘要】BACKGROUND: The limited viability of mesenohymal slim cells restricts its application in treatments vivo. Therefore.it is veryimportant to find in experiment method to prolong the viability of mesenchymjlstem cells so as to lay foundation for experimentvivo.OBJECT WE: To construct the lentiral vector encoding survin gene of mice with clone vectorpLV.Des3d.P/hygro.METHODS: Oligo nucleic acid software was used to design a couple of primers according to survivin gene sequence of micewhich is publicated on Genbark The attB recombination site was added to the two ends of the couple of primers, and mSurvinandIRES/EmGFP were amplified by PCR. The PCR product liter BP reaction ?as transferred to Stb13. And the correctpDomn-mSurvivin ind pTail-IRES/EmGFP were selected and sequenced. The mixture of pD own-mSuvition. pTail RESSWSFPand pLV.Des3d.Pmygro was used to do LR reaction. The lentiviral vector pLV.EX3d.P/hygro-EF1A>mSurvvin>IRESEmGFP Wasamplified and sequenoed again.RESULTS AND CONCLUSION: The results of PCR ind sequencing showed that the lenfei.al vector encoding survivin gene ofmice mis constructed successfulty, which lays foundation tor the further study of survr?in anti-apoptosis inmesenchymalstemcells.%背景:间充质干细胞体内有限的存活能力严重制约其用于体内治疗方面的应用,因此寻找一种能延长存活能力的实验方法是间充质干细胞进行体内实验的重要前提.目的:应用pLV.Des3d.P/hygro构建小鼠survivin基因的慢病毒表达载体.方法:采用Oligo核酸软件对Genbank上所发表的小鼠survivin mRNA序列进行分析,设计一对survivin基因上下游引物,利用Gateway 克隆方法,分别在其两端添加attB重组位点,运用PCR扩增mSurvivin和IRES/EmGFP,将PCR产物进行BP反应然后转化到Stbl3感受态菌,通过卡那霉素抗性筛选、PCR鉴定,选取鉴定正确的pDown-mSurvivin和pTail-IRES/EmGFP 克隆测序.将pDown-mSurvivin和pTail-IRES/EmGFP与pLV.Des3d.P/hygro 混合进行LR反应,构建慢病毒载体pLV.EX3d.P/hygro-EF1A>mSurvivin>IRES/EmGFP,再次进行PCR、测序的鉴定.结果与结论:显示基因序列克隆正确,成功构建了含有小鼠survivin基因的重组慢病毒表达载体.说明慢病毒表达载体pLV.EX3d.P/hygro-EF1A>mSurvivin>IRES/EmGFP构建成功,为下一步研究survivin在间充质干细胞中抗凋亡作用奠定基础.【总页数】5页(P9244-9248)【作者】杨平;苟欣;周青松;何卫阳;余昆【作者单位】重庆医科大学附属第一医院泌尿外科,重庆市,400016;重庆医科大学附属第一医院泌尿外科,重庆市,400016;重庆医科大学附属第一医院泌尿外科,重庆市,400016;重庆医科大学附属第一医院泌尿外科,重庆市,400016;重庆医科大学附属第一医院泌尿外科,重庆市,400016【正文语种】中文【中图分类】R394.2【相关文献】1.小鼠Stra8基因慢病毒表达载体的构建与鉴定 [J], 郑哲;郑英2.Gateway技术构建小鼠血管生成素-1慢病毒表达载体及其病毒包装 [J], 李秋平;马新娜;张小英;许靖;章晟;王春枝;封志纯3.小鼠Smad4基因慢病毒表达载体构建及在293T细胞中的表达 [J], 孙毓晗;侯昭晖;肖玉丽4.小鼠S1PR3慢病毒表达载体的构建及表达 [J], 王航;蔡克银;黄浩5.小鼠survivin基因重组真核表达载体的构建及鉴定 [J], 余昆;苟欣;杨华安;周青松;夏阳因版权原因,仅展示原文概要,查看原文内容请购买。

动物医学进展,2021 ,2(1)69-74ProgressinVeterinary Medicine靶向SynDIG1的shRNA 慢病毒载体的构建及功能鉴定李亚琳,史秀超,权美平(渭南师范学院环境与生命科学学院,陕西渭南714099)摘 要:为了构建靶向突触分化诱导基因1(SynDIG1 )的shRNA 慢病毒表达载体,设计出SynDIG1的 siRNA 的靶点序列,并合成含干扰序列的双链DNA 发卡结构shRNA,合成的Oligo 经过退火分别与双酶切处理后的pLKO. 1-GFP 载体连接。
将构建的重组质粒pLKO. 1-GFP-SynDIG1 shRNA 转化到DH5a 感受态细菌中,过夜培养后挑选阳性克隆子,扩增后提取DNA 进行质粒测序,得到两个序列正确的重组质粒。
用这些重组质粒转染HEK293T 细胞以及大鼠海马神经元细胞,蛋白免疫印迹及细胞免疫荧光检测SynDIG1 shRNA 对外 源性和内源性SynDIG1表达的敲减作用。
进一步用重组质粒转染HEK293T 细胞产生慢病毒颗粒,用病毒颗粒 感染DIV2和DIV14的神经细胞,免疫印迹检测SynDIG1的表达。
结果表明这两个重组质粒均能够有效地抑制外源性和内源性SynDIG1的表达,对HEK293T 中瞬时表达的SynDIG1敲减率达到75%,其慢病毒颗粒感染对早期神经细胞中内源性SynDIG1的表达抑制也很明显。
用pLKO. 1-GFP 成功构建了能够有效抑制外源性 和内源性SynDIG1表达的重组质粒,为探讨RNA 干扰技术抑制神经细胞SynDIG1基因表达的相关研究奠定基础,为研究SynDIG1基因在神经传导和突触发育及其可塑性调节中的作用提供了有力工具。
关键词:SynDIG1 ;慢病毒;shRNA ;基因敲减技术中图分类号:S852. 615;Q789突触分化诱导基因(synapse differentiation in duced gene1,SynDIG1)是一种高度保守的跨膜蛋 白,在中枢神经系统的兴奋性突触传递和突触可塑性调节中起着重要的作用[1]。



现物医学 Progress in Modern Biomedicine V〇L20 NO^l NOV*2020• 4007 •doi: 10.13241/ki.pmb.2020.21.002CX3CL1 RN A干扰慢病毒载体的构建包装与鉴定*柏秀娟郭艳蛾时霄冰孙博吴卫平A(中国人民解放军总医院第二医学中心神经内科北京100853)摘要目的:构建包装趋化因子C X3C的配体1(CX3CL1)RNA干扰慢病毒栽体。
方法:参考目的基因CX3CL1序列,设计P C R引物扩增相应的干扰序列,然后将干扰序列连接至pLVX-shRNA2酶切后的线性化栽体上,通过酶切及测序鉴定获得阳性克隆,即为构建成功的pLVX-shRNA2-CX3CLl慢病毒干扰栽体。
将构建好的慢病毒干扰载体同慢病毒包装栽体共同转染293T细胞,收 集上清,纯化浓缩后即为pLVX-shRNA2-CX3CLl慢病毒。
最后将慢病毒感染B M SC s细胞,QPCR检测慢病毒的干扰效率结果:经过酶切能切出大小约为6500bp和1350bp的两条带,获得与预期结果相一致的D N A片段,并通过测序验证了序列的准确性,成功构建CX3CL1 R N A慢病毒干扰栽体。
然后经过包装、纯化浓缩后得到pLVX-shRNA2-CX3CLl慢病毒UQPCR结果表明干扰 组明显抑制了C X3C Llm R N A的表达,干扰效率在70%以上。
结论:成功构建包装CX3CL1干扰慢病毒栽体,并证实其显著沉默 了BM SCs细胞C X3C L1的表达,为CX3CL1在BM SCs移植的大鼠缺血性脑卒中炎症反应的机制研究奠定基础。
关键词:C X3C L1;R N A干扰;骨髄间充质干细胞;缺血性脑卒中;慢病毒中图分类号:R-33;Q78;R743文献标识码:A文章编号:1673-6273(2020 )214007-06Construction, Packaging and Identification of CX3CL1 RNAiLentiviral Vector*BAIXiu-juan, GUO Yan-e, SHI Xiao-bing, SUN Bo, WU Wei-pin^(Neurological Department o f t he Second Medical Center, Chinese PLA General Hospital, Beijing, 100853, China) ABSTRACT Objective: To construct and pack CX3CL1 RNAi lentiviral vector. Methods: With reference to the target gene CX3CL1 sequence, PCR primers were designed to amplify the corresponding interference sequence. Then the interference sequence was connected to pLVX-shRNA2 linearized vector.Positive clones were obtained by identification o f PCR and digestion, which was a successful construction o f pLVX-shRNA2-CX3CLl Lentiviral interference vectors. The constructed RNAi lentiviral vector was transfected into 293T cells together with the packaging vectors. The supernatant was collected, purified and concentrated to pLVX-shRNA2-CX3CLl lentivirus. Finally, BMSCs were infected with lentivirus, and the interference efficiency was detected by QPCR. Results: After digestion, two bands with 6500bp and 1350bp were obtained consistent with the expected results, and the sequence accuracy was verified by sequencing. The CX3CL1 RNAi lentiviral vector was successfully constructed.After packaging, purification and concentration, the pLVX-shRNA2-CX3CLl lentivirus was obtained. QPCR results showed that the interference group significantly inhibited the expression o f CX3CLlmRNA, and the interference efficiency was above 70%. Conclusion: The CX3CL1 RNAi lentiviral vector was successfully constructed and packed and significantly silenced the expression o f CX3CL1 o f BMSCs, which laid the foundation for the study o f the mechanism o f CX3CL1 on inflammatory response of ischemic stroke in BMSCs transplanted rats.Keywords: CX3CL1; RNA interference; BMSCs; Ischemic stroke; LentivirusChinese Library Classification(CLC): R-33; Q78; R743 Document code: AArticle ID: 1673-6273(2020)21-4007-06刖目缺血性脑卒中是导致人类残疾、死亡的重要疾病之一是由血栓或栓塞导致血管阻塞引起脑组织缺血、缺氧,进一步 可致脑组织神经元发生不可逆性死亡,同时启动了缺血局部组 织的非特异性炎症反应,过度的炎症反应可进一步加剧脑组织 的继发性损伤。
∗基金项目:北京市自然科学基金资助项目(编号:7192084);首都卫生发展科研专项(编号:2020-2-1152);北京市属医学研究所公益发展改革试点项目(就医研2021-10)作者单位:100069北京市首都医科大学附属北京佑安医院北京肝病研究所第一作者:霍云飞,男,23岁,硕士研究生㊂E-mail:yunfeihuo@ 共同第一作者:寇卜心,女,32岁,大学本科,检验技师㊂E-mail:koubuxin@通讯作者:石英,E-mail:yingshi@ ㊃实验性肝炎㊃体外靶向TP53BP2基因shRNA慢病毒载体的构建及功能鉴定∗霍云飞,寇卜心,柴梦音,豆双双,高明慧,石英,刘晓霓㊀㊀ʌ摘要ɔ㊀目的㊀本研究旨在构建靶向肿瘤蛋白p53结合蛋白2(TP53BP2)基因的短发夹RNA(shRNA)慢病毒载体,以抑制肝癌细胞TP53BP2的表达㊂方法㊀设计了2对针对TP53BP2基因的RNA干扰序列,并合成相应的shRNA序列㊂shRNA退火形成双链oligo序列后,应用基因重组技术构建重组质粒,经菌落PCR和测序鉴定,将重组正确的质粒进行慢病毒包装和滴度测定,并采用Western Blot㊁qRT-PCR和激光共聚焦技术观察慢病毒Lenti-shTP53BP2对HepG2细胞TP53BP2基因的干扰效果㊂结果㊀测序比对结果显示,各重组慢病毒载体与设计参考序列一致,提示各重组慢病毒体构建成功;重组慢病毒载体经慢病毒包装后,显示pHS-ASR-LW429㊁pHS-ASR-LW512和pHS-ASR-LW513的滴度分别为9.7ˑ108TU/mL㊁6.1ˑ108TU/mL和6.4ˑ108TU/mL;用慢病毒Lenti-shTP53BP2(pHS-ASR-LW512和pHS-ASR-LW513)感染HepG2细胞后,与对照慢病毒(pHS-ASR-LW429)比,经Western Blot㊁qRT-PCR和激光共聚焦结果显示两个Lenti-shTP53BP2均能显著下调HepG2细胞TP53BP2基因水平和蛋白表达量㊂结论㊀本研究成功构建了靶向TP53BP2基因shRNA慢病毒载体,其能有效下调HepG2细胞TP53BP2的表达,为进一步研究TP53BP2在肝癌发生发展过程中的机制研究奠定了基础㊂㊀㊀ʌ关键词ɔ㊀HepG2细胞;肿瘤蛋白p53结合蛋白2;短发夹RNA;慢病毒;体外㊀㊀DOI:10.3969/j.issn.1672-5069.2023.02.004㊀㊀Construction and functional verification of a shRNA lentiviral vector targeting to TP53BP2gene in HepG2cells in vitro㊀Huo Yunfei,Kou Buxin,Chai Mengyin,et al.Beijing Institute of Hepatology,You an Hospital,Capital Medical University, Beijing100069,China㊀㊀ʌAbstractɔ㊀Objective㊀The present paper aimed to inhibit the expression of tumor suppressor p53-binding protein2 (TP53BP2)in liver cancer by short hairpin RNAs(shRNAs)with lentiviral vector.Methods㊀Two pairs of RNA interference sequences targeting to TP53BP52gene were designed,and their corresponding shRNA sequences were synthesized.After annealing of shRNA to form double-stranded oligo sequences,the recombinant plasmid was constructed by gene recombination technique. The correct recombinant plasmid was used after PCR and sequencing identification of the colony for lentivirus packaging and titer determination.The interference effect of lentivirus lenti-shTP53BP2on TP53BP2gene in HepG2cells was observed by Western Blot,qRT-PCR and laser confocal technique.Results㊀The sequencing alignment results showed that each recombinant lentiviral vector was consistent with the designed reference sequence,indicating that each recombinant lentiviral vector was successfully constructed;the titers of pHS-ASR-LW429,pHS-ASR-LW512and pHS-ASR-LW513were9.7ˑ108TU/mL,6.1ˑ108TU/mL and6.4ˑ108TU/mL,respectively;the HepG2cells were infected with lentivirus lenti-shTP53BP2(pHS-ASR-LW512and pHS-ASR-LW513),and the results of Western blot,qRT-PCR and laser confocal technique showed that the two lenti-shTP53BP2significantly down-regulated the TP53BP2RNA level and itsprotein expression in HepG2cells as compared with the controllentivirus(PHS-ASR-LW429).Conclusion㊀In this study,we successfully construct the shRNA lentiviral vector targeting toTP53BP2gene with the capacity of effectively down-regulation ofTP53BP2expression in HepG2cells,which might lay afoundation for further research on the mechanism of TP53BP2inthe hepatocarcinogenesis.㊀㊀ʌKey wordsɔ㊀HepG2cells;Tumor suppressor p53-binding protein2;Short hairpin RNA;Lentivirus;In vitro㊀㊀据2019年世界卫生组织统计,恶性肿瘤是大多数国家70岁前死亡的第一或第二大主要原因[1]㊂肝癌作为最常见的恶性肿瘤之一,在2020年统计研究时显示其新发病例数和死亡病例数分别位居第六位和第三位,依然存在复发率高和生存率低的问题[1-3]㊂肿瘤抑制蛋白p53结合蛋白2(tumor sup-pressor p53-binding protein2,TP53BP2),又称p53凋亡刺激蛋白2(apoptosis stimulating protein2of p53,ASPP2),是一个由TP53BP2基因编码的全长为1128个氨基酸的肿瘤抑制因子[4]㊂在其C端, TP53BP2具有一个富含脯氨酸结构域(Pro),四个锚蛋白结构域(Ank)和一个SH3结构域;在其N端, TP53BP2具有一个泛素样折叠结构(ULD)和一个α-螺旋结构[5]㊂TP53BP2表达与肿瘤的发生发展有着密切的关系㊂研究显示,胃癌[6]㊁乳腺癌[7,8]㊁子宫内膜癌[9]㊁口腔癌[10]等多种恶性肿组织TP53BP2呈低表达,而TP53BP2表达与肝癌的发生发展的关系日益得到关注㊂RNA干扰(RNA interference, RNAi)是指在进化过程中高度保守的双链RNA诱发的基因沉默现象,能特异性干扰靶基因的表达[11]㊂慢病毒载体能稳定介导基因沉默,具有转染效率高和稳定表达的优势[12]㊂本研究旨在构建靶向TP53BP2基因的短发夹RNA(short hairpin RNA, shRNA)慢病毒载体,以抑制肝癌细胞TP53BP2的表达,为后续TP53BP2在肝癌发生发展中的机制研究奠定基础㊂1㊀材料与方法1.1质粒㊁慢病毒载体㊁细胞与试剂㊀质粒小量快速提取试剂盒(Aidlab公司);限制性内切酶(Thermo 公司);慢病毒载体pLV-hU6-shRNA-hef1a-mNeon-green-P2A-Puro购自北京合生基因科技有限公司㊂HepG2细胞和293T细胞均由本实验室保存㊂T4 DNA ligase㊁慢病毒包装试剂盒㊁EpFect Transfection Reagent㊁EvaGreen2ˑMaster Mix(Syngen Tech公司);FastPure Cell/tissue Total RNA Isolation Kit V2 (Vazyme公司);PrimeScriptTM II1ST Strand cDNA Synthesis Kit㊁TB Green Premix Ex TaqTM(Takara公司);彩虹180广谱蛋白Marker(Solarbio公司); DMEM培养基㊁胎牛血清和Puromycin(Gibco公司);抗β-actin单克隆抗体(Cell Signaling Technology公司);抗TP53BP2单克隆抗体(Abcam公司)㊂1.2目的基因干扰靶点的设计㊀根据美国国家生物技术信息中心(National Center for Biotechnology In-formation,NCBI)提供的TP53BP2基因的核酸序列,设计siRNA靶向序列和阴性对照序列(表1)㊂表1㊀siRNA靶向序列siRNA㊀㊀靶向序列TP53BP2shRNA01GGATCTGACTCTTGCTGAACT TP53BP2shRNA02GCTGAGAATCAGGAAGCTAA NC shRNA03AAACGTGACACGTTCGGAGAA 1.3shRNA的合成及退火㊀shRNA由北京合生基因科技有限公司进行合成㊂在合成的shRNA中,loop 茎环结构为CGAA(表2)㊂将合成的单链shRNA用oligo annealing buffer溶解成20μM,互补单链各取30μl进行混合,置于95ħ水浴锅中加热5min,然后室温冷却,形成双链oligo片段㊂取双链oligo片段1μl用于后续的连接反应,其余置于-20ħ保存㊂表2㊀TP53BP2基因沉默慢病毒载体信息载体编号载体内容shRNA序列pHS-ASR-LW512TP53BP2shRNA015 -GGATCTGACTCTTGCTGAACT CGAAAGTTCAGCAAGAGTCAGATCC-3 pHS-ASR-LW513TP53BP2shRNA025 -GCTGAGAATCAGGAAGCTAAG CGAACTTAGCTTCCTGATTCTCAGC-3 pHS-ASR-LW429NC shRNA035 -AAACGTGACACGTTCGGAGAA CGAATTCTCCGAACGTGTCACGTTT-31.4载体质粒与退火的shRNA连接㊀取载体质粒pLV-hU6-shRNA-hef1a-mNeongreen-P2A-Puro,经BsaI酶切线性化后与退火的双链oligo序列在表3的连接反应体系下于16ħ过夜连接,完成慢病毒基因沉默质粒构建(图1)㊂其中,阳性对照所加的退火的双链oligo是经验证连接效果较好的片段,与连接组所加的退火的双链oligo长度一样,但与目的序列无关㊂表3㊀连接反应体系试剂阳性对照(μL)自连对照(μL)连接组(μL)退火的双链oligo10mM1 1线性化的干扰载体40ng/μL333 10ˑT4DNA ligase Buffer222 T4DNA ligase111 dd H2O Up to20Up to20Up to20图1㊀TP53BP2基因沉默慢病毒质粒示意图1.5转化㊁菌落鉴定与测序㊀用构建完成的质粒转化DH5α感受态细胞,然后涂布于含有氨苄西林(ampi-cillin,Amp)抗性的LB固体培养基平板,37ħ培养过夜㊂经培养后挑取单一菌落进行菌落PCR筛选阳性克隆,对得到的阳性克隆进行测序验证(测序引物序列:CAGGAAGAGGGCCTATTTCCC)㊂对经测序验证正确的阳性克隆,用质粒小量快速提取试剂盒提取质粒,取提取出的质粒用于慢病毒的包装㊂1.6慢病毒包装㊀取3~5ˑ106个293T细胞,接种于10cm细胞培养皿中,置于37ħ,5%CO2的培养箱中过夜培养㊂然后,使用慢病毒包装试剂盒中的包装质粒混合物(Package Plasmid Mix)和EpFect Transfection Reagent等试剂进行慢病毒包装系统转染㊂在转染48h后,收集含有慢病毒的上清液,更换新鲜培养基继续培养24h,再次收集上清㊂将收集到的上清和浓缩试剂按照5:1的比例在4ħ过夜进行混合㊂混合后,在4ħ,4000g条件下离心30min,收集沉淀㊂使用PBS重悬沉淀,即得到病毒浓缩液㊂将收集到的病毒浓缩液分装,保存于-80ħ㊂1.7慢病毒滴度测定㊀取1ˑ105个293T细胞,接种于6孔板,置于37ħ,5%CO2的培养箱中过夜培养,用于病毒滴度测定㊂每种病毒分别按0.1μL㊁1μL和10μL体积接种3个孔,并添加终浓度为8μg/mL的促感染试剂Polybrene㊂培养72h后拍照,记录病毒感染后的情况并提取基因组DNA,用于qRT-PCR检测㊂qRT-PCR实验的扩增/检测对象为慢病毒载体上的WPRE序列(WPRE序列可以随目的基因整合入细胞基因组)㊂根据公式TU ml-1= (CˑNˑDˑ1000)/V,即可计算出慢病毒滴度㊂其中,C为平均每基因组整合的病毒拷贝数;N为感染时细胞的数目;D为病毒载体的稀释倍数,V为加入的稀释病毒的体积数(μL)㊂1.8慢病毒转染HepG2细胞和干扰效果鉴定㊀取1ˑ105个HepG2细胞,接种于6孔板,过夜培养后,在孔中加入慢病毒和8μg/mL的Polybrene㊂每孔所加病毒量(μL)=MOIˑ感染时的细胞数/病毒滴度ˑ1000,其中HepG2的MOI值为10~30㊂转染24h 后更换新鲜培养基,继续培养24~48h,拍照,记录感染情况,并加入嘌呤霉素筛选转染成功的细胞㊂收集转染成功的HepG2细胞,分别采用Western blot㊁qRT-PCR和激光共聚焦技术观察TP53BP2基因在蛋白质和mRNA水平上的表达情况㊂GAPDH 和TP53BP2引物序列见表4㊂表4㊀qPCR引物序列名称序列GAPDH F:GGAGCGAGATCCCTCCAAAATR:GGCTGTTGTCATACTTCTCATGGASPP2F:ATTCGTCTTTGGAGGGAGAR:ATTGTGAAGAGCCGTGATG1.9统计学方法㊀计量资料以xʃs表示,采用Student s t检验,应用SPSS24.0软件进行分析,P< 0.05为差异有显著性统计学意义㊂2㊀结果2.1TP53BP2基因沉默慢病毒质粒载体构建成功㊀我们将退火的双链oligo序列与经BsaI酶切割后的线性化载体在T4DNA ligase的作用下进行连接,并经菌落PCR筛选阳性克隆和DNA测序,测序比对结果显示各重组质粒载体序列与设计序列一致(图2)㊂由此可见,pLV-hU6-shTP53BP2-hef1a-mNeongreen-P2A-Puro质粒(pHS-ASR-LW512; pHS-ASR-LW513)构建成功㊂图2㊀TP53BP2基因沉默慢病毒质粒测序比对结果A:pHS-ASR-LW429;B:pHS-ASR-LW512;C:pHS-ASR-LW513 2.2慢病毒Lenti-shTP53BP2包装和滴度检测情况㊀我们采用慢病毒包装试剂盒和293T细胞包装基因沉默慢病毒质粒,48h后在荧光显微镜下观察到所有细胞均具有GFP绿色荧光,说明慢病毒包装成功(图3)㊂提取经慢病毒转染后的293T细胞基因组DNA,进行qRT-PCR检测,经数据分析得出慢病毒的滴度,其中pHS-ASR-LW429为9.66ˑ108TU/ mL,pHS-ASR-LW512为6.13ˑ108TU/mL,pHS-ASR-LW513为6.36ˑ108TU/mL(表5㊁表6和表7)㊂2.3Lenti-shTP53BP2对HepG2细胞干扰效果情况㊀我们使用Lenti-shTP53BP2(pHS-ASR-LW512和pHS-ASR-LW513)和对照Lenti-NC shRNA(pHS-ASR-LW429)转染HepG2细胞24h后,更换新鲜培养基继续培养24~48h,添加含有6μg/mL嘌呤霉素的培养基进行筛选㊂当未转染慢病毒的细胞在含有嘌呤霉素的培养基中全部死亡后,转入2μg/mL 嘌呤霉素的培养基维持筛选,所有细胞表达GFP绿色荧光,提示转染成功(图4A)㊂经Western blot㊁qRT-PCR和激光共聚焦技术观察HepG2细胞TP53BP2表达,发现在mRNA水平和蛋白质水平上pHS-ASR-LW512和pHS-ASR-LW513两种慢病毒感染的HepG2细胞TP53BP2表达均显著降低(图4 B~E)㊂表5㊀pHS-ASR-LW429滴度测定项目V值(μL)C值N值D值滴度(TU/mL)平均滴度(TU/mL) 11047.22ˑ10519.4ˑ10821 5.12ˑ1051 1.0ˑ1099.7ˑ108 30.10.52ˑ10519.4ˑ108表6㊀pHS-ASR-LW512滴度测定项目V值(μL)C值N值D值滴度(TU/mL)平均滴度(TU/mL) 11032.62ˑ1051 6.5ˑ10821 3.42ˑ1051 6.9ˑ109 6.1ˑ108 30.10.32ˑ1051 5.0ˑ108表7㊀pHS-ASR-LW513滴度测定项目V值(μL)C值N值D值滴度(TU/mL)平均滴度(TU/mL) 11028.42ˑ1051 5.7ˑ10821 3.32ˑ1051 6.6ˑ109 6.4ˑ108 30.10.32ˑ1051 6.8ˑ1083㊀讨论TP53BP2作为一个重要的肿瘤抑制基因,在肿瘤,尤其是肝癌的发生发展过程中发挥着重要作用㊂研究表明,TP53BP2参与自噬[13]㊁细胞增殖[14]㊁化疗耐药性[15,16]等信号的调节,影响着肝癌的发生发展和患者预后㊂我们先前的研究也发现,敲除TP52BP2基因可促进二乙基亚硝胺(DEN)诱导的小鼠肝癌的发生[17]㊂本研究针对TP53BP2基因设计合成了2条RNA干扰寡核苷酸链,采用基因重组技术和慢病毒包装技术成功构建了靶向TP53BP2基因shRNA慢病毒载体㊂进一步利用成功构建的Lenti-shTP53BP2慢病毒转染HepG2细胞,经Western blot㊁qRT-PCR和激光共聚焦技术观察其靶向沉默图3㊀TP53BP2基因沉默慢病毒包装过程中239T 细胞GPF 绿色荧光表达情况(48h)图4㊀Lenti -shTP53BP2对HepG2细胞的干扰效果A:荧光显微镜观察HepG2细胞经慢病毒感染后GFP绿色荧光表达(100ˑ);B:激光共聚焦观察HepG2细胞经慢病毒感染后TP53BP2蛋白表达;C /D:Western blot 验证HepG2细胞经慢病毒感染后TP53BP2蛋白表达;E:qRT -PCR 验证HepG2细胞经慢病毒感染后TP53BP2mRNA 水平与pHS -ASR -LW429比,∗P <0.05,∗∗P ∗<0.001TP53BP2基因的效果㊂结果发现无论是从蛋白水平还是mRNA 水平,pHS -ASR -LW512和pHS -ASR -LW513两种慢病毒均能有效抑制HepG2细胞TP53BP2的表达㊂实验的成功为进一步研究TP53BP2基因在肝癌发生发展过程中的机制奠定了基础㊂ʌ参考文献ɔ[1]Sung H,Ferlay J,Siegel R L,et al.Global cancer statistics 2020:GLOBOCAN estimates of incidence and mortality worldwide for 36cancers in 185countries.Ca Cancer J Clin,2021,71(3):209-249.[2]Tian G,Yang S,Yuan J,et parative efficacy of treatmentstrategies for hepatocellular carcinoma:systematic review and network meta -analysis.BMJ Open,2018,8(10):e21269.[3]Kulik L,El -Serag H B.Epidemiology and management of hepato-cellular carcinoma.Gastroenterology,2019,156(2):477-491.[4]Samuels -Lev Y,O'Connor D J,Bergamaschi D,et al.ASPP pro-teins specifically stimulate the apoptotic function of p53.Mol Cell,2001,8(4):781-794.[5]Ahn J,Byeon I L,Byeon C H,et al.Insight into the structural ba-sis of pro -and antiapoptotic p53modulation by ASPP proteins.JBiol Chem,2009,284(20):13812-13822.[6]Gen Y,Yasui K,Kitaichi T,et al.ASPP2suppresses invasion andTGF -beta1-induced epithelial -mesenchymal transition by inhibiting Smad7degradation mediated by E3ubiquitin ligase ITCHin gastric cancer.Cancer Lett,2017,398:52-61.[7]Wu T,Song H,Xie D,et al.Mir -30b -5p proliferation,migration,and invasion of breast cancer cells via targeting ASPP2.Biomed Res Int,2020,2020:7907269.[8]Wu T,Song H,Xie D,et al.Silencing of ASPP2promotes theproliferation,migration and invasion of triple -negative breast cancercells via the PI3K /AKT pathway.Int J Oncol,2018,52(6):2001-2010.[9]Konno T,Kohno T,Okada T,et al.ASPP2suppression promotesmalignancy via LSR and YAP in human endometrial cancer.Histo-chem Cell Biol,2020,154(2):197-213.[10]Patel K D,Barasiya Y V,Patel J B,et al.Apoptosis stimulatingprotein of p53(ASPP)1and ASPP2m -RNA expression in oral cancer.Arch Oral Biol,2020,119:104920.[11]Mondal M,Klimov P,Flynt A S.Rewired RNAi -mediated genomesurveillance in house dust mites.PLoS Genet,2018,14(1):e1007183.[12]Poling B C,Tsai K,Kang D,et al.A lentiviral vector bearing a re-verse intron demonstrates superior expression of both proteins andmicroRNAs.RNA Biol,2017,14(11):1570-1579.[13]Chen R,Wang H,Liang B,et al.Downregulation of ASPP2im-proves hepatocellular carcinoma cells survival via promoting BECN1-dependent autophagy initiation.Cell Death Dis,2016,7(12):e2512.[14]Liang B,Chen R,Song S,et al.ASPP2inhibits tumor growth byrepressing the mevalonate pathway in hepatocellular carcinoma.Cell Death Dis,2019,10(11):830.[15]Xu L,Tong X,Zhang S,et al.ASPP2suppresses stem cell -likecharacteristics and chemoresistance by inhibiting the Src /FAK /Snail axis in hepatocellular carcinoma.Tumour Biol,2016,37(10):13669-13677.[16]Yang T,Gao Y,Liu D,et al.ASPP2enhances chemotherapeuticsensitivity through the down -regulation of XIAP expression in a p53independent manner in hepatocellular carcinoma.Biochem Biophys Res Commun,2019,508(3):769-774.[17]Wang S,Kou B,Chai M,et al.Knockout of ASPP2promotes DEN-induced hepatocarcinogenesis via the NF -kappaB pathway inmice.Cancer Gene Ther,2022,29(2):202-214.(收稿:2022-10-14)(本文编辑:陈从新)。
第36卷第2期暨南大学学报(自然科学与医学版)Vol.36 No.22015年4月JournalofJinanUniversity(NaturalScience&MedicineEdition)Apr. 2015基于Cre LoxP系统的新型慢病毒表达载体pLOX CMV E/P的构建与鉴定高文勇1, 夏景波2,3, 陈卓莹2,3, 吴彩红2,3, 王健欢2,3, 龙颖妍2,3, 齐绪峰2,3(1.武汉市汉口医院神经内科,湖北武汉430012;2.暨南大学再生医学教育部重点实验室,广东广州510632;3.暨南大学生命科学技术学院发育与再生生物学系,广东广州510632)[摘 要] 目的:基于Cre LoxP系统构建携带EGFP及Puromycin抗性基因,并带有巨细胞病毒(CMV)及翻译延伸因子1α(EF1α)双启动子的新型慢病毒表达载体,为基因治疗及基因功能研究提供有效的慢病毒载体系统.方法:以已经插入LoxP序列的pLOX TERT iresTK慢病毒载体为模板,用SpeI与KpnI进行双酶切,去除TERT iresTK片段,然后与人工设计合成的多克隆位点片段连接,构建pLOX MCS表达载体.同时,以pB513载体为模板扩增EF1α EGFP Puro表达框(带有BamHI及KpnI酶切位点),然后与经过BamHI及KpnI双酶切的pLOX MCS载体连接,进而构建pLOX CMV EF1α EGFP Puro(简称pLOX CMV E/P)载体.将pLOX CMV E/P载体与慢病毒包装载体pCMVR8.74及pMD2.G共转染293T细胞,包装病毒进行报告基因的功能分析.结果:菌落聚合酶链式反应(PCR)、酶切鉴定及测序结果均与预期结果一致,绿色荧光蛋白及抗药性基因均有较好的活性与功能.结论:成功构建了pLOX MCS及pLOX CMV E/P慢病毒表达载体,为基因治疗及基因功能研究提供有效的慢病毒表达系统.[关键词] 慢病毒表达载体; Cre LoxP系统; 绿色荧光蛋白基因; 嘌呤霉素抗性基因[中图分类号] Q782;Q784;Q786 [文献标志码] A [文章编号] 1000-9965(2015)02-0160-08doi:10.11778/j.jdxb.2015.02.013ConstructionandverificationofnovellentiviralexpressionvectorpLOX CMV E/PbasedonCre LoxPsystemGAOWenyong1, XIAJingbo2,3, CHENZhuoying2,3, WUCaihong2,3,WANGJianhuan2,3, LONGYingyan2,3, QIXufeng2,3(1.DepartmentofNeurology,HankouHospitalofWuhan,Wuhan430012,China;2.KeyLaboratoryofRegenerativeMedicineofMinistryofEducation,JinanUniversity,Guangzhou510632,China;3.DepartmentofDevelopmentalandRegenerativeBiology,CollegeofLifeScienceandTechnology,JinanUniversity,Guanghzou510632,China)[Abstract] Aim:ToconstructanovellentiviralexpressionvectorbasedontheCre LoxPsystem.Thenovellentiviralexpressionvectorconsistsoftowreportergenes(EGFPandpuromycinresistancegene(Puro)drivenbyEF1αpromoter,andthemultipleclonesite(MCS)drivenbyCMVpromoter).Meth ods:pLOX TERT iresTKlentiviralvectorwasusedasthetemplateanddigestedbySpeIandKpnIre[收稿日期] 2014-10-23[基金项目] 国家自然科学基金项目(81270183,81100079,81211140351);广东省自然科学基金项目(S2013010013598);教育部留学回国人员科研启动基金项目(2013-693);中央高校基本科研业务费专项资金项目(11614601);广州市珠江科技新星专项(2014J2200002).[作者简介] 高文勇(1977-),男,医师,研究方向:神经内科学通信作者:齐绪峰,副教授,硕士生导师,Tel:020-85222687;E-mail:qixufeng@jnu.edu.cnstrictionenzymestoremovetheTERT iresTKfragment.ThelinedvectorwasthenligatedwithanovelMSCfragmentincludingSpeIandKpnIcohesiveendsiteswhichwasgotbyannealingtwooligonucleoti des,therebyconstructingpLOX MCSvector.Atthesametime,EF1α EGFP PurofragmentwithBamHIandKpnIcohesiveendsiteswasamplifiedfromthepB513plasmidandligatedwithpLOX MCSvectordi gestedbyBamHIandKpnIrestrictionenzymes,therebyconstructingpLOX CMV EF1α EGFP Puro(pLOX CMV E/P)vector.pLOX CMV E/Pvectorwasco transfectedinto293TcellswithpackagingplasmidsincludingpCMVR8.74andpMD2.Gtoexaminethevirustitersandfunctionofreportergenes.Results:Throughsequencinganalysis,itwasfoundthatthesequenceofpLOX CMV E/Pvectoriscor rect.Furthermore,thefunctionofreportergenes(EGFPandPuromycin)wasverifiedbyfluorescencemicroscopeandpuromycintreatment,respectively.Conclusion:Takentogether,thisstudyhassuccess fullyconstructedanovellentiviralvectorpLOX CMV E/PincludingtworeportergenesdrivenbyEF1αpromoterandaMCSdrivenbyCMVpromoter,whichprovidesamoreeffectivelentiviralexpressionsys temforgenetherapyandfunctionstudies.[Keywords] Lentiviralexpressionvector; Cre LoxPsystem; EGFP; Puromycinresistancegene 慢病毒载体是以人类免疫缺陷病毒(humanim mune deficiencyvirus,HIV)为基础改造而来,具有宿主范围广,能感染分裂相细胞及非分裂相细胞,转染效率高,能携带多达8~10kb的外源基因等优势,是基因功能研究及基因治疗领域的重要途径之一[1-2].近年来,随着分子生物学技术的深入发展,根据慢病毒载体的物理特性将其分装到几个不同的质粒中,进一步提升了慢病毒载体用于基因治疗的安全性.这种技术改进不但使得慢病毒载体所携带的外源基因有效整合到宿主细胞的基因组并稳定表达,同时又具有较低的免疫原性,进而能有效降低慢病毒载体作为基因治疗手段的潜在风险[3].而且,与逆转录病毒载体相比,慢病毒载体通过随机整合的方式将外源基因插入到宿主细胞的基因组中,因此不容易造成原癌基因及致癌基因位点的插入突变[4].目前,基于慢病毒载体的基因转染已经在造血干细胞、自然杀伤(NK)细胞、树突状细胞(DC)细胞等免疫细胞中取得成功,从而为有关免疫细胞相关疾病的基因治疗提供了一条有效途径[5-6].而且,慢病毒载体系统在肿瘤基因治疗领域同样显示出良好的应用前景[7].2006年,Levine等进行了第一项基于慢病毒载体的人体临床实验,利用表达靶向HIV的反义基因的慢病毒载体转染T细胞,并回输到患者体内,用于治疗5名经过多个疗程的抗病毒治疗仍然无效的HIV患者.结果表明,其中4名患者体内观察到靶向HIV的细胞免疫,在随后的4年随访中,未发现白血病或其他严重的与基因治疗相关的不良反应,进一步表明了慢病毒载体在临床基因治疗中基本安全[8].虽然通过慢病毒载体能将外源基因有效整合到靶细胞的基因组中实行稳定表达,但外源基因及慢病毒载体骨架序列的整合对靶细胞仍然存在潜在的安全风险,无法根据需要将外源基因及时删除,急需研发出既能有效筛选、鉴定转染的阳性细胞,又能根据需要有效删除外源基因及载体骨架的更加安全有效的慢病系统.位点特异性基因重组技术是通过对特定的DNA序列进行精确切割及连接,实现在基因水平上对生物体进行遗传改造的一种基因操作技术,其中Cre LoxP系统已经发展成为一种成熟且广泛应用的删除目的基因片段的方法.本研究首先利用已经在3′端LTR区域的U3原件中插入了LoxP位点的慢病毒载体pLOX TERT riesTK为基础,通过设计、合成并插入多克隆位点(MCS)构建pLOX MCS载体,同时在人工合成的MCS末端引入由EF1α启动子驱动的EGFP绿色荧光蛋白基因及Puromycin抗性基因(Puro)片段,进而构建既能进行荧光示踪及抗药性筛选,又能根据需要适时外源基因的新型慢病毒表达载体pLOX CMV EF1α EGFP Puro(简称pLOX CMV E/P),为基因治疗及基因功能研究提供一种安全有效的慢病毒载体系统.1 材料与方法1.1 材料(1)质粒与菌株 pLOX TERT iresTK(12245)、pCMVR8.74(22036)、pMD2.G(12259)及pX458(48138)质粒购自美国Addgene公司.DH5α感受态购自北京天根生化科技有限公司(货号:CB101).PiggyBac转座子质粒pB513B 1购自于美国System161 第2期高文勇,等:基于Cre LoxP系统的新型慢病毒表达载体pLOX CMV E/P的构建与鉴定Biosciences(SBI)公司.(2)试剂 DMEM细胞培养基(DulbeccoModifiedEagleMedium,DMEM)(货号:SH30243.01B)购自于Hyclone公司,胎牛血清(货号:10099-141)、Opti MEM IReducedSerumMedium(货号:31985-062)购自Gibco公司,限制性内切酶、T4DNA连接酶(货号:EL0014)及T4PNK(货号:EK0031)均购自于Thermo公司,高保真酶KOD Plus Neo(货号:KOD 401)购自日本TOYOBO公司,分子量标准DL5000Marker(货号:3428A)、DL15000Marker(货号:DL10003)分别购自日本Takara公司和上海捷瑞生物工程有限公司,QIAprep SpinMiniprepKit质粒提取试剂盒(货号:27104)购自于美国Qiagen公司,TIANgelMidiPurificationKit凝胶回收试剂盒(货号:DP209)购自北京天根生化科技有限公司,AxyPrepTMPCRCleanupKit纯化试剂盒购自Axygen公司(货号:AP PCR 50),脂质体转染试剂LipoFiterTM(货号:HB TRLF 1000)购自汉恒生物科技(上海)有限公司,嘌呤霉素(Puromycin)(货号:P8833)购自美国Sigma公司.慢病毒浓缩试剂盒(货号:V2001 02)够购自美国BIOMIGA公司.2×Taq NeoPCRMasterMix(货号:M0201)购自美津生物技术有限公司.SOCOutgrowthMedium(货号:B9020)购自NewEnglandBiolabs公司.氨苄青霉素(Ampicillinsodiumsalt)(货号:BS050B)购自BIO SHARP公司.1.2 方法(1)pLOX MCS载体构建 以pLOX TERT ir esTK质粒DNA为模板,利用NEBcutterV2.0(ht tp://tools.neb.com/NEBcutter2/)在线分析软件进行限制性酶切位点分析,选择质粒中没有的常用限制性酶切位点的DNA序列,构建人工多克隆位点序列:设计合成MCS F:5′ CTAGTGATCAGAATTCTTAATTAACTCGAGTCGACGGATCCTGCAGGACGCGTGGTAC 3′及MCS R:5′ CACGCGTCCTGCAGGATCCGTCGACTCGAGTTAATTAAGAATTCTGATCA 3′引物对,退火形成双链,构成多克隆位点序列(图1).反应体系:MCS F(100μM)1μL,MCS R(100μM)1μL,T4PNK0.5μL,10×T4LigaseBuffer1μL,补足ddH2O至10μL.退火条件:37℃30min,95℃5min,25℃pause(0 08℃/s降温).将pLOX TERT iresTK质粒经SpeI和KpnI双酶切,切胶回收8195bp大小片段,并与上述退火形成的MCS片段进行连接,用T4DNA连接酶于16℃酶连过夜,重组质粒中MCS上游的CMV启动子中的测序引物(CMV F:5′ CGCAAATGGGCGGTAGGCGTG 3′)进行测序(北京六合华大基因科技有限公司),测序正确的重组质粒命名为pLOX MCS.(2)pLOX CMV E/P载体构建 以pB513质粒为模板,设计正向引物EF1α GFP/Pur F:5′ CGGGATCCGAAGGATCTGCGATCGCTCC 3′(下划线为BamHI酶切位点),以及反向引物EF1α GFP/Pur R:5′ GGGGTACCTCAGGCACCGGGCTTGCGGGT 3′(下划线为KpnI酶切位点),用KOD Plus Neo高保真酶扩增EF1α EGFP Puro片段,大小为1986bp,切胶回收目的大小片段.PCR体系为:10×PCRBuffer(5μL),2mMdNTPs(5μL),25mMMgSO4(3μL),上下游引物(10μM)各1 5μL,pB513质粒0 3μL(约50ng),高保真酶1μL,补足ddH2O至50μL.PCR条件为:94℃预变性2min,98℃10s,58℃退火30s,68℃延伸1min10s;共35个循环;最后于68℃延伸7min.PCR产物用质量分数为1%的琼脂糖凝胶电泳进行检查.pLOX MCS载体与PCR产物经BamHI和KpnI双酶切,切胶纯化后进行酶连反应.然后转化感受态大肠杆菌DH5α,涂布于含有氨苄青霉素(质量浓度100μg/mL)的LB平板,37℃培养过夜.重组质粒经过菌落PCR挑选阳性克隆及双酶切鉴定后,分别利用CMV F及Sp6 R(5′ ATTTAGGTGACACTATAG 3′)进行正、反双向测序,CMV F正向测序引物位于插入位点上游的CMV启动子内部,Sp6 R反向测序引物位于3′LTR区域(测序由北京六合华大基因科技有限公司完成).测序正确的重组质粒命名为pLOX CMV EF1α EGFP Puro(pLOX CMV E/P),并提取质粒备用.(3)pLOX CMV E/P载体表达绿色荧光蛋白及Puromycin抗性基因的功能验证 将本研究所构建成功的慢病毒表达载体pLOX CMV E/P(或pLOX MCS)分别与包装质粒pCMVR8.74及包膜质粒pMD2.G共转染293T细胞,进行病毒颗粒的包装.将293T细胞提前一天接种到10cm的培养皿中,待第2天达到70%~80%融合时进行上述表达载体与包装载体的共转染,转染1d后观察293T细胞绿色荧光表达情况.转染体系:pLOX CMV E/P(或pLOX MCS)载体:pCMVR8 74质粒:pMD2.G质粒的摩尔比为1 5∶1∶1,三者的总DNA量为24μg,与LipoFiterTM脂质体转染试剂共转染293T细胞,转染48h后收集上清液,同时加入10mL新鲜培养基并于转染后72h后收集上清,将两次收集的病毒上清261暨南大学学报(自然科学与医学版)第36卷液混合,3000r/min水平离心去除细胞碎片,上清液通过Milipore0 45μm滤膜过滤,进一步去除细胞碎片.将过滤后的病毒上清液与慢病毒浓缩试剂盒(BIOMIGA,美国)中的LPbuffer按照4∶1(V/V)的比例混合,并于4℃过夜.然后将病毒混合液于4℃条件下,以3000r/min的转速离心30min,移除上清,用慢病毒浓缩试剂盒中的LSbuffer(100μL)溶解病毒颗粒沉淀,并于4℃条件下,以8000rpm/min的转速离心2min,收集上清液,分装后于-80℃冻存备用.将293T细胞提前一天种于24孔板中,待细胞融合到50%~60%时用5μL慢病毒颗粒进行感染,3天后加入嘌呤霉素(Puromycin),终质量浓度为3μg/mL,继续培养至4天及7天,用荧光显微镜检测细胞存活及绿色荧光蛋白表达情况.只进行慢病毒感染,而不进行Puromycin处理的细胞直至感染7天后观察绿色荧光蛋白表达情况;未经任何处理的细胞用作空白对照.2 结果2.1 pLOX MCS载体的构建及测序鉴定pLOX MCS载体的构建流程如图1所示,本研究所设计合成的人工多克隆位点包含SpeI、BclI、EcoRI、PacI、XhoI、SalI、BamHI、SbfI、MluI及KpnI等10个酶切位点.将所挑选的重组质粒进行测序鉴定,筛选到与预期序列一致的重组质粒,命名为pLOX MCS(图2).所插入的MCS位点位于CMV启动子下游,可用于插入外源基因片段.A:pLOX TERT iresTK载体图谱;B:合成的人工多克隆位点图谱;C:pLOX MCS载体图谱.图1 pLOX MCS慢病毒表达载体构建流程Fig.1 SchematicrepresentationoftheestablishmentofpLOX MCSvector图2 pLOX MCS载体的测序结果分析Fig.2 SequencinganalysisofpLOX MCSvector2.2 pLOX CMV E/P载体的构建及鉴定pLOX CMV EF1α EGFP Puro载体(pLOX CMV E/P)的构建流程如图3所示,其多克隆位点包含SpeI、BclI、EcoRI、PacI、XhoI、SalI及BamHI等7个酶切位点,其上游为CMV启动,下游为EF1α启动子驱动的EGFP及Puromycin抗性基因.以pB513载体为模板进行PCR扩增,获得1986bp大小的片段,与预期结果一致(图4A).将目的大小片段切胶回收,经BamHI及KpnI双酶切并纯化,然后与经BamHI及KpnI双酶切的pLOX MCS载体进行连接反应,转化大肠杆菌,挑选5个单菌落进行菌落PCR361 第2期高文勇,等:基于Cre LoxP系统的新型慢病毒表达载体pLOX CMV E/P的构建与鉴定A:pLOX MCS载体图谱;B:pB513载体图谱;C:pLOX CMV E/P载体图谱.图3 pLOX CMV EF1αEGFP Puro(pLOX CMV E/P)载体的构建流程Fig.3 SchematicrepresentationoftheestablishmentofpLOX CMV EF1αEGFP Puro(pLOX CMV E/P)vector A:EF1α EGFP Puro片段的PCR扩增.M为DNA分子量,Exp为实验组的PCR产物;B:重组子的菌落PCR验证.M为DNA分子量,1-5号泳道分别为挑选的5个单克隆.图4 PCR扩增及重组子的菌落PCR验证Fig.4 PCRamplificationandtheverificationofrecombinantsbycolonyPCRanalysis检测,结果发现1#与5#克隆扩增出与预期大小一致的目的片段(图4B).将1#与5#克隆提取的质粒用BamHI及KpnI进行双酶切鉴定,两个克隆均检测出1986bp及8225bp两条与预期大小一致的目的片段(图5A).对其中的1#克隆插入位点的上、下游分析进行正、反双向测序分析,结果表明:以位于CMV启动子内部的正向测序引物CMV F测序的结果与部分CMV启动子、MCS及EF1α启动子的5′端DNA序列完全一致(图5B);而且,以位于3′LTR内部的反向测序引物Sp6 R测序的结果与部分3′LTR、KpnI酶切位点及Puro mycin抗性基因编码区的C端DNA序列完全一致(图5C).461暨南大学学报(自然科学与医学版)第36卷A:1号与5号克隆的双酶切(BamHI及KpnI)鉴定结果;B:1号克隆的正向测序结果;C:1号克隆的反向测序结果.图5 重组载体的酶切鉴定及序列分析Fig.5 Enzymedigestionandsequencinganalysisofrecombinant2.3 报告基因功能鉴定慢病毒载体与包装质粒共转染293T细胞1天后,荧光显微镜观察结果表明:用pLOX CMV E/P表达载体与包装质粒共转染靶细胞后,能够产生稳定的绿色荧光,表明绿色荧光蛋白能够稳定表达;然而,用pLOX MCS载体与包装质粒共转染靶细胞后,未观察到绿色荧光(图6).上述结果表明所构建的pLOX CMV E/P慢病毒表达载体中EGFP能够有效工作.同时,用Puromycin(3μg/mL,经筛选发现此浓度下处理4天能全部杀死正常的293T细胞)处理慢病毒感染的293T细胞,结果表明:未处理的正常细胞能够有效存活,但无绿色荧光蛋白表达;经慢病毒感染而未经Puromycin处理的细胞仍然观察到大量存活的细胞,并且可观察到相当比例的细胞中表达绿色荧光蛋;然而,用Puromycin处理慢病毒感染的细胞4天后,仅有少量细胞存活,且存活的细胞均能表达绿色荧光蛋白;而且,继续用Puromycin筛选到7天后,仍然有存活并能表达绿色荧光蛋白的细胞(图7).该结果表明,所构建的慢病毒表达载体pLOX CMV E/P中的嘌呤霉素抗性基因及EGFP编码基因能够协同表达,同时发挥各自的生物学功能.图6 重组载体pLOX CMV E/P中EGFP报告基因的功能验证Fig.6 IdentificationofEGFPexpressedbypLOX CMV E/Pvectorbyfluorescencemicroscopy561 第2期高文勇,等:基于Cre LoxP系统的新型慢病毒表达载体pLOX CMV E/P的构建与鉴定图7 重组载体pLOX CMV E/P中Puromycin抗性基因的功能验证Fig.7 IdentificationofpuromycinresistancegeneexpressedinpLOX CMV E/Pvectorbyfluorescencemicroscopy3 讨论 随着分子生物学技术的不断发展,基因治疗被认为是治愈疾病的有效方式之一.基因治疗面临的主要问题是如何将治疗基因有效转移到靶细胞及靶组织中,使其以适当的水平表达而发挥疾病治疗功能.因此,基因传递系统中载体的选择十分关键,它不但决定了外源基因的转染效率,同时还决定了外源基因表达的靶向性及有效性[9].目前,用于基因传递的病毒载体主要包括逆转录病毒、腺病毒及慢病毒载体等几种类型.其中,逆转录病毒载体虽然能够将外源基因整合到宿主细胞的基因组实现稳定表达,但不能转染非分裂相细胞,且在干细胞中的表达效率较低;腺病毒载体虽然能够同时转染分裂相及非分裂相细胞,但目的基因不能整合到靶细胞的基因组中,仅能实现瞬时表达[10-11].而慢病毒载体同时克服了上述两种载体的缺陷,具有容量大、转染效率高、细胞毒性小、可长期稳定表达外源基因、免疫原性低等优势,已经成为基因治疗领域的重要途径[12].目前,国际上已开展了多项基于改造后的慢病毒载体作为基因递送工具的临床试验,从而给一些因基因缺陷而导致的致命性疾病患者带来了希望.用于基因治疗及基因功能研究的理想的慢病毒载体不仅能够有效将外源基因稳定地整合到宿主细胞的基因组中,同时应当带有合适的示踪及筛选标记,以满足体外筛选及体内示踪的研究需要.更重要的是,如能利用特定的分子生物学手段,根据需要适时将整合到宿主基因组中的外源基因及筛选与示踪标记进行有效删除,可有效提高慢病毒载体系统在临床应用领域的安全性.Cre重组酶是源于P1噬菌体cre基因所编码的一个Int家族蛋白,可特异性识别基因组上由34个核苷酸序列构成的LoxP位点,并能根据LoxP位点的方向性实现两个LoxP位点之间DNA序列的有效删除、插入及倒置,从而实现不同的基因编辑功能.而且,Cre LoxP系统对靶基因的编辑无需其他辅助因子的参与,同时没有种属特异性.Cre LoxP位点特异性重组系统因其具有作用方式简单,且可利用特异性启动子实现在生物体发育的特定时间及特定组织中表达,已经广泛应用于基因的功能鉴定及基因组的修饰[13].因此,本研究利用Cre LoxP系统的工作原理,构建既能适时删除外源基因及载体原件,又能对靶细胞实行有效筛选及荧光示踪的慢病毒表达载体,以便为基因治疗及基因功能研究提供一种安全有效的慢病毒载体系统.本研究构建的pLOX CMV MCS EF1αEGFP PuroR(pLOX CMV E/P)慢病毒表达载体带有绿色荧光蛋白基因(EGFP)及嘌呤霉素抗性基因(Puro)661暨南大学学报(自然科学与医学版)第36卷两个报告基因,既能实现荧光示踪,又能实现抗性筛选;而且两个报告基因均在广谱性启动子EF1α驱动下表达,两者之间用T2A连接肽进行融合,不但能实现两种报告基因的高水平表达,又能使两者的表达水平互不影响.本研究构建的慢病毒载体同时带有多克隆位点,用于外源基因的插入,其上游带有另一种广谱性启动子CMV,用于驱动外源基因的表达.同时,本慢病毒表达载体框架的3′端LTR区域的构成原件(U3 R U5)已经进行了遗传改造,已经将LTR区域内U3原件-418至-18之间的碱基序列删除掉,保留U3原件上游35个碱基及下游18个碱基,并在中间插入了LoxP序列,进而实现在慢病毒载体3′端LTR区域的U3原件内插入LoxP序列[14].通过慢病毒载体的转录及反转录过程,最终在外源基因及表达原件整合到宿主基因中的同时,在其两端同时整合了同向的LoxP位点,在Cre重组酶表达的情况下,包含外源基因在内的整合片段将被有效删除.因此,利用本研究构建的慢病毒载体,结合Cre重组酶在不同时间及不同细胞类型中的表达情况,能够实现外源基因的条件性敲除,为更加精确地研究外源基因的功能提供了有力工具.本研究所构建的慢病毒表达载体pLOX CMV E/P可用慢病毒包装质粒pCMVR8.74及慢病毒包膜质粒pMD2.G进行病毒颗粒的包装,本实验利用所构建的新型慢病毒表达载体及上述慢病毒包装载体共转染293T细胞,通过慢病毒浓缩试剂进行病毒颗粒浓缩,感染293T细胞3天后能够稳定表达绿色荧光蛋白.同时,用3μg/mL的Puromycin连续筛选4天后,野生型293T细胞全部死亡,而用含有pLOX CMV E/P载体包装的慢病毒颗粒转染的靶细胞不但能够存活,并且稳定表达绿色荧光蛋白,表明本研究所构建的慢病毒表达载体pLOX CMV E/P能够有效工作.本研究成功构建的慢病毒表达载体pLOX CMV E/P不但能为基因功能研究提供一种有效的基因操作工具,同时有望为下一步的基因治疗研究提供一种安全高效的慢病毒系统.[参考文献][1] CASESS,PRICEMA,JORDANCT,etal.StabletransductionofquiescentCD34(+)CD38(-)humanhematopoieticcellsbyHIV 1 basedlentiviralvectors[J].ProcNatlAcadSciUSA,1999,96(6):2988-2993.[2] CHANGLJ,GAYEE.Themoleculargeneticsoflenti viralvectors———currentandfutureperspectives[J].CurrGeneTher,2001,1(3):237-251.[3] KUNGSK,ANDS,BONIFACINOA,etal.Inductionoftransgene specificimmunologicaltoleranceinmyeloablatednonhumanprimatesusinglentivirallytransducedCD34+progenitorcells[J].MolTher,2003,8(6):981-991.[4] MONTINIE,CESANAD,SCHMIDTM,etal.Hemato poieticstemcellgenetransferinatumor pronemousemodeluncoverslowgenotoxicityoflentiviralvectorintegration[J].NatBiotechnol,2006,24(6):687-696.[5] MAHMOODS,KANWARN,TRANJ,etal.SHP 1phosphataseisacriticalregulatorinpreventingnaturalkillercellself killing[J].PLoSOne,2012,7(8):e44244.[6] TRANJ,MAHMOODS,CARLYLEJR,etal.AlteringthespecificityofNK:targetcellinteractionsbygeneticmanipulationofNKreceptorexpressiononprimarymouseNKcells[J].Vaccine,2010,28(22):3767-3772.[7] UPRETID,PATHAKA,KUNGSK.Lentiviralvector basedtherapyinheadandneckcancer(Review)[J].OncolLett,2014,7(1):3-9.[8] LEVINEBL,HUMEAULM,BOYERJ,etal.Genetransferinhumansusingaconditionallyreplicatinglentiviralvector[J].ProcNatlAcadSciUSA,2006,103(46):17372-17377.[9] OTTO WILHELMMOHAMED,MOHAMEDAL RUBEAI.Viralvectorsforgenetherapy[M].NewYork:SpringerSicence+BusinessMedia,2011:183-209.[10]VANDENDRIESSCHET,VANSLEMBROUCKV,GOO VAERTSI,etal.Long termexpressionofhumancoagulationfactorVIIIandcorrectionofhemophiliaAafterinvivoretroviralgenetransferinfactorVIII deficientmice[J].ProcNatlAcadSciUSA,1999,96(18):10379-10384.[11]CONNELLYS,SMITHTA,DHIRG,etal.InvivogenedeliveryandexpressionofphysiologicallevelsoffunctionalhumanfactorVIIIinmice[J].HumGeneTher,1995,6(2):185-193.[12]VIGNAE,NALDINIL.Lentiviralvectors:excellenttoolsforexperimentalgenetransferandpromisingcandidatesforgenetherapy[J].JGeneMed,2000,2(5):308-316.[13]张文娟,于丹妮.Cre/loxP位点特异性重组系统及其在口腔医学领域中的应用[J].国际口腔医学杂志,2011,38(1):55-58.[14]SALMONP,OBERHOLZERJ,OCCHIODOROT,etal.Reversibleimmortalizationofhumanprimarycellsbylentivector mediatedtransferofspecificgenesc.MolTher,2000,2(4):404-414.[责任编辑:陈咏梅]761 第2期高文勇,等:基于Cre LoxP系统的新型慢病毒表达载体pLOX CMV E/P的构建与鉴定。
人核糖核酸酶抑制因子siRNA慢病毒载体的构建及鉴定丁宁;邱景剑;王定友;李劭恒;张云;李坤【期刊名称】《大连大学学报》【年(卷),期】2016(37)6【摘要】本实验旨在构建人核糖核酸酶抑制因子稳定干涉载体pLKO.1-dsRI,为后续观测人核糖核酸酶抑制因子干涉载体的表达对肿瘤细胞的影响奠定基础.用亚克隆法,将针对人核糖核酸酶抑制因子的干涉片段从Simple T-dsRI质粒克隆到pLK-0.1-TRC质粒,用酶切法筛选得到阳性重组质粒pLKO.1-dsRI,用测序法鉴定克隆序列正确.转染时设干扰组、空载体组和空白组三组,每组三次重复.用脂质体CellfectinR将pLKO.1-dsRI(干扰组)、pLKO.1-TRC(空载体组)转染进人肝癌细胞HepG2细胞中,未处理的细胞作空白组.转染后72hr用Western blotting检测细胞中核糖核酸酶抑制因子表达的变化.双酶切鉴定及测序鉴定结果均正确;Western blotting结果表明,对比空白组(1.1578±0.015)和空载体组(1.1216±0.027),干扰组RI表达(0.6119±0.048)明显下调(P<0.05).结果表明人核糖核酸酶抑制因子干涉载体pLKO.1-dsRNH1构建成功.【总页数】4页(P62-65)【作者】丁宁;邱景剑;王定友;李劭恒;张云;李坤【作者单位】大连大学医学院,辽宁大连 116622;大连大学医学院,辽宁大连116622;大连大学医学院,辽宁大连 116622;大连大学医学院,辽宁大连 116622;大连大学医学院,辽宁大连 116622;大连大学医学院,辽宁大连 116622【正文语种】中文【中图分类】Q782【相关文献】1.靶向人核糖核酸酶抑制因子双抗性的 shRNA 逆转录病毒载体构建及鉴定 [J], 李坤;丁宁2.绿色荧光蛋白融合人核糖核酸酶抑制因子逆转录病毒载体pLNCX-EGFP-C1-hri 的构建及鉴定 [J], 李坤;李琳;王福强;田余祥;曲淑贤;崔秀云3.沉默HepG2细胞核糖核酸酶抑制因子的shRNA逆转录病毒载体的构建及鉴定[J], 李琳;李坤;王福强;丁宁4.人核糖核酸酶抑制因子基因重组腺病毒载体的构建及鉴定 [J], 刘鹏;赵宝昌;樊建慧;田余祥;杨帆;崔秀云5.重组人核糖核酸酶抑制因子基因的siRNA表达载体构建及B16细胞中基因沉默的鉴定 [J], 李坤;田余祥;陈海波;杨帆;樊建慧;赵宝昌;崔秀云因版权原因,仅展示原文概要,查看原文内容请购买。
逆转录病毒载体pOZ—KLF5的构建及鉴定摘要:为研究KLF5的相互作用蛋白从而深入探索其生物功能,构建了C端带有FLAG、HA、6xHis三标签的pOZ-KLF5真核表达逆转录病毒栽体。
pOZ-KLF5栽体可通过一次转录得到双顺反子转录单位,即一个转录物能表达两种独立存在的蛋白质:三标签融合蛋白KLF5-3xTag和筛选标记——白细胞介素-2受体a (IL-2Ra)。
通过偶联IL-2Ra抗体的磁珠成功筛选出KLF5-3xTag表达阳性的293T细胞,经Western-blot检测细胞内KLF5-3xTag表达情况及其对293T细胞增殖及周期的影响,发现KLF5-3xTag能够促进细胞克隆的形成并使S期细胞增多,且可被已知E3泛素连接酶WWP1降解,与已有报道一致。
进一步通过免疫沉淀实验证明了与KLF5融合的标签的免疫反应性。
该质粒的成功构建为研究KLF5相互作用蛋白及深入探索其生物功能奠定了基础。
关键词:KLF5;双顺反子转录;真核表达;三标签融合蛋白中图分类号:Q812 文献标识码:A 文章编号:1007-7847(2015)02-0124-07Construction and Characterizations of pOZ-KLF5 Retroviral PlasmidZHAO Xin-yu,WANG Wei-xi,LU Ze-lan,HUANG Lei,ZHAO Ke-wen*(Department of Pathophysiology,Shanghai Jiao Tong University School of Medicine,Shanghai 200025,China)Abstract:To analyze KLF5 interacting proteins and explore the biological functions of KLF5,the eukaryotic expression retroviral plasmid pOZ-KLF5,with the FLAG,HA,6xHis triple tags at C-terminal,was con?structed. Plasmid pOZ-KLF5 can get a bicistronic transcription unit after once transcription and express two independent proteins:triple-tagged fusion protein KLF5-3xTag and selection marker interleukin-2 receptor a (IL-2Ra). 293T cells expressing KLF5-3xTag were successfully selected by the magnetic beads coupling IL-2Ra antibody,and the expression of KLF5-3xTag was detected by Western-blot. Then,the proliferation and cell cycle of 293T were tested,and the results indicated that KLF5-3xTag promotes the proliferation of cell colonies and the Gl/S transition.In addition,co-expression of WWP1,a known E3 ligase of KLF5,can decrease KLF5-3xTag protein level,which is consistent with previous reports. Furthermore,immunoprecipi- tation experiment was applied to prove the immunoreaction of the tags. The successful construction of pOZ- KLF5 provides a powerful tool to study the interacting proteins of KLF5 and to explore the biological func?tions of KLF5.Key words:KLF5;bicistronic transcription;eukaryotic expression;triple-tagged fusion protein(Life Science Research,2015,19(2):124?130)KLF5蛋白是类KtlippeKKriippel-likefactors,KLF)转录因子家族的一员,属于基础转录因子。
到冃前为止该家族巳经有17个成员被鉴定出来。
KLF5又名BETB2(basictranscriptionelement-bindingprotein2)和IKLF (intestine-enrichedKriippel-likefactor),在胚胎发育、细胞周期、细胞生长与分化、维持细胞干性等生物过程中发挥作用。
研究发现KLF5在人和小鼠的小肠、结肠、胃、胰腺、胎盘、睾丸、骨骼肌、肺、膀胱和子宫等组织中广泛表达[1-3]。
KLF5能与多个基因的GCbox区域结合从而调控其转录。
KLF5蛋白C端含有KLFs家族特征性的、可以结合基因组DNA的3个串联重复的锌指(zincfinger,ZF)结构域,在ZF结构域前含有富含脯氨酸的转录激活结构域。
值得关注的是,KLF5在不同的细胞类型中发挥的作用不同,已有的研究提示这可能与KLF5的翻译后修饰有关。
KLF5蛋白有不同的翻译后修饰,包括磷酸化、乙酰化、泛素化和SUMO化修饰,经过翻译后修饰的KLF5可以与不同的蛋白相互作用从而发挥不同甚至完全相反的功能[4.5]。
为了研究KLF5在真核生物不同生理及病理条件下的相互作用蛋A从而深入研究其生物学功能,我们构建了逆转录病毒真核表达载体P0Z-KLF5,通过磁珠筛选出阳性克隆,在检测KLF5生物功能的同时,验证了与目的蛋白KLF5融合的标签的免疫反应性。
1材料与方法1.1材料大肠杆菌DH5a感受态(Invitrogen,美国),pOZ-C载体[6](简称pOZ,C端带有FLAG和HA标签,Addgene,美国),质粒提取试剂盒(QIAGEN,美国),凝胶回收试剂盒、PCR纯化试剂盒(Tian-gen,中国),XhoⅠ、NotI核酸内切酶、T4DNA连接酶(Takara,日本),LB培养基(Oxoid,英国),X-tremeGENE9转染试剂(Roche,美国),MG132(Sig-ma,美国),抗FLAG-M2亲和凝胶、抗HA亲和凝胶(Sigma,美国),抗IL-2Ra抗体(Upstate,美国),Ni-NTA琼脂糖(QIAGEN,美国),DynabeadsM450(Invitrogen,美国),抗KLF5抗体(Protein-Tecli,美国)c1.2pOZ-KLF5表达载体的构建根据逆转录病毒载体pOZ 的多克隆位点,设计带有XhoI和NotI酶切位点(见斜体)的KLF5扩增引物,在C端PCR引物中引人了6xHis标签(见下划线)。
引物序列Forward:5’(-GGGCrCGAGTGCCACCATGGCTACAAGGG-TGCTGAGC-3’,Reverse5’-CCCGCGGCGGCQI-GGTGGTGGTGGTGGTGGTTCTGGTGCCTCTTCA-TATG -3’。
以人KLF5的CDNA为模板,PCR扩增C末端带有6xHis标签的KLF5基因’命名为ALF5-6xHis。
PCR条件如下:94℃变性3min,94°C 30s,55℃30s,72℃1min,30个循环,72℃;延伸10min,4℃保存。
PCR产物经1%琼脂糖凝胶电泳,利用凝胶回收试剂盒回收PCR产物,然后分别用XhoI和I酶切PCR 产物和P0Z-C载体,用胶回收试剂盒回收酶切后的AXF5片段与载体,用T4DNA 连接酶将KLF5-6xHis与pOZ载体连接,转化感受态DH5a,挑选阳性克隆于3mL 的LB培养基小量扩增,并用质粒提取试剂盒提取所构建的质粒。
经XhoI和Not Ⅰ酶切确定目的片段的插入,测序证实插入载体目的片段的正确性,质粒被命名为P0Z-KLF5。
1.3稳定表达pOZ-KLF5质粒的细胞系筛选采用Roc.he公司的X-tremeGENE9试剂将逆转录病毒载体P0Z-KLF5连同病毒包装辅助质粒gag-Pol、VSV-g—起转染包装细胞,48h后收集具有感染能力的逆转录病毒感染293T细胞,由于P0Z-KLF5质粒可以同时转录并表达两种蛋白质:目的蛋白KLF5和筛选标记白细胞介素-2受体a(IL-2Ra),因此可以通过偶联IL-2Ra抗体的DynabeadsM450磁珠筛选pOZ-KI,F5病毒感染的293T细胞,从而得到稳定表达三标签融合的目的蛋白KLF5(简称KLF5-3xTag)的293T细胞系。
1.4pOZ-KLF5质粒在真核细胞表达的鉴定收集1.3中获得的稳定表达KLF5-3xTag的293T细胞的总蛋白,WestemBlot方法检测目的蛋白KLF5-3xTag的表达情况。
1.5细胞水平检测KLF5的生物学功能使用1.3中获得的稳定表达KLF5-3xTag的293T细胞进行克隆形成实验和细胞周期分析实验,检测KLF5-3xTag蛋白的表达对细胞增殖及细胞周期的影响。
克隆形成实验分别将100个 1.3筛选的对照组和KLF5-3xTag表达组293T细胞接种到六孔板中培养10d,吸去上清液,用PBS 洗两遍,冰甲醇固定细胞后结晶紫染色,晾干后拍照,并对直径超过100μm的克隆进行统计分析。
使用同样的对照组与KLF5-3xTag表达组293T细胞检测细胞周期:分别离心收集lxlO6个细胞,用PBS洗两遍后,加入预冷70%乙醇于-20°C 固定过夜,第二天用PBS洗两遍,加入PI染料和RNaseA于37℃避光孵育30min,最后用流式细胞仪检测周期。
1.6Western-blot检测WWP1对KLF5-3xTag蛋白的降解在1.3中获得的稳定表达KLF5-3xTag的293T细胞内共转染WWP1质粒(美国Emory大学DrnigJT教授惠赠)或者对照载体,收集细胞前4h加入20μmol/LMG132处理,收集细胞总蛋白Westem-blot检测细胞中KLF5蛋白的含量变化,β-actin作为内参。