HPLC法测定紫草中的左旋紫草素的含量
- 格式:doc
- 大小:30.00 KB
- 文档页数:3

·238·紫草ZicaoRADIX ARNEBIAE本品为紫草科植物新疆紫草Arnebia euchroma(Royle)Johnst.或内蒙紫草Arnebia guttata Bunge的干燥根。
春、秋二季采挖,除去泥沙,干燥。
【性状】新疆紫草(软紫草) 呈不规则的长圆柱形,多扭曲,长7~20cm,直径1~2.5cm。
表面紫红色或紫褐色,皮部疏松,呈条形片状,常10余层重叠,易剥落。
顶端有的可见分歧的茎残基。
体轻,质松软,易折断,断面不整齐,木部较小,黄白色或黄色。
气特异,味微苦、涩。
内蒙紫草呈圆锥形或圆柱形,扭曲,长6~20cm,直径0.5~4cm。
根头部略粗大,顶端有残茎1或多个,被短硬毛。
表面紫红色或暗紫色,皮部略薄,常数层相叠,易剥离。
质硬而脆,易折断,断面较整齐,皮部紫红色,木部较小,黄白色。
气特异,味涩。
【鉴别】(1)取本品粉末0.5g,置试管中,将试管底部加热,生成红色气体,并于试管壁凝结成红褐色油滴。
(2)取本品粉末0.5g,加石油醚(60~90℃)20ml,超声处理20分钟,滤过,滤液浓缩至约1ml,作为供试品溶液。
另取紫草对照药材0.5g,同法制成对照药材溶液。
照薄层色谱法(附录ⅥB)试验,吸取上述两种溶液各4µl,分别点于同一硅胶G薄层板上,以环己烷-甲苯-乙酸乙酯-甲酸(5:5:0.5:0.1)为展开剂,展开,取出,晾干。
供试品色谱中,在与对照药材色谱相应的位置上,显相同的紫红色斑点;再喷以10%氢氧化钾甲醇溶液,斑点变为蓝色。
【检查】水分照水分测定法(附录Ⅸ H第一法)测定,不得过15.0%。
【含量测定】羟基萘醌总色素取本品适量,于50℃干燥3小时,粉碎(过三号筛),精密称取0.5g,置100ml量瓶中,加乙醇至刻度,4小时内时时振摇,滤过。
精密量取续滤液5ml,置25ml量瓶中,加乙醇至刻度,摇匀。
照紫外-可见分光光度法(附录Ⅴ A),在516nm波长处测定吸光度,按左旋紫草素(C16H16O5) )为242计算,即得。

广东地区市售紫草紫草素含量的抽样检测
黄卫战;张继平
【期刊名称】《基层中药杂志》
【年(卷),期】1994(008)001
【摘要】紫草始载于《神农本草经》。
祖国医学认为它有凉血、活血、解毒透疹的作用,常用于血热毒盛、斑疹紫黑、麻疹不透、疮疡、温疹、水火烫伤等症。
紫草中紫草素为其疗效的主要成分,含量越高质量越好,与其质量有着直接关系。
为了保证紫草质量,《中国药典》1990版.一部,制定了紫草中紫草素含量测定标准。
云南、新疆等地据此标准抽查了本地市售紫草的合格率,但广东地区市售紫草质量的合格率尚未见报道。
【总页数】2页(P21-22)
【作者】黄卫战;张继平
【作者单位】不详;不详
【正文语种】中文
【中图分类】R284.1
【相关文献】
1.RP-HPLC法同时测定新疆紫草中紫草素、乙酰紫草素、β,β-二甲基丙烯酰紫草素的含量 [J], 孙健文;温宪春;蔡德富;王玥;吴永会
2.HPLC法测定不同产地紫草中去氧紫草素的含量 [J], 朱永乐;董娜;曲晓兰;齐永秀
3.紫草油及紫草素油的含量测定方法研究 [J], 沈丽宫; 叶棣柏; 罗明可; 尤洁; 何文胜
4.紫草总色素β-环糊精包合物的定性鉴别及左旋紫草素含量测定 [J], 李俊江; 刘向娥; 李季文; 张增志; 牛建雄
5.不同产地蒙紫草中左旋紫草素和β,β'-二甲基丙烯酰阿卡宁含量测定及质量标准研究 [J], 吕晓洁;渠弼;刘德旺;程丹丹;那生桑
因版权原因,仅展示原文概要,查看原文内容请购买。

新疆紫草中紫草素提取方法研究张继业;杜蕊;刘婷婷;杨范莉;童华【摘要】Objective To optimize the extraction of shikonin from A rnebia euchroma with solvent extraction in four solvents .Meth-ods The extractions were carried out in petroleum ether ,ethanol ,chloroform ,and 10 g ・ L - 1 NaOH solution with stirring at 25 ℃ ,solvent extraction time of 30 min ,and solid to liquid ratio 1 ∶ 9 .The extraction process was conducted in duplicate .The shikonin content in extracts were measured by HPLC with gradient elution .Extraction methods were evaluated by extraction yield .Results The yield of shikonin extraction differed in four solvents (10 g ・ L - 1 NaOH solution > ethanol > chloroform > petro-leum ether) .Conclusion The shikonin extraction in NaOH solution provides higher extraction yield and significantly reduces the amount of organic solvent .This economic and environmental method is suitable for shikonin extraction from A rnebia enchroma .%目的:采用石油醚、乙醇、氯仿、碱液4种溶剂提取紫草素,选出最优提取方法。

高效液相色谱法测定紫草油中β,β'-二甲基丙烯酰阿卡宁的含量王贵英;骆勇;左宏笛【期刊名称】《中国医院用药评价与分析》【年(卷),期】2013(013)012【摘要】目的:建立紫草油中β,β'-二甲基丙烯酰阿卡宁的含量测定方法.方法:采用高效液相色谱(HPLC)法,色谱柱为迪马C18钻石色谱柱(250 mm×4.6mm,5μm);流动相为乙腈-水-甲酸(75∶25∶0.2);检测波长为275 nm;柱温为室温.结果:在80.6 ~564.5 ng范围内,β,β'-二甲基丙烯酰阿卡宁进样量和峰面积具有良好的线性关系,线性回归方程为C=1.027 9×10-6A +2.358 9×10-2,r =0.999 9.测得紫草油中β,β'-二甲基丙烯酰阿卡宁平均含量为0.869 2 mg/ml.结论:该方法简便可行,重现性好,可作为紫草油的质量控制方法.【总页数】3页(P1089-1091)【作者】王贵英;骆勇;左宏笛【作者单位】贵州省食品药品检验所,贵阳550004;贵州省食品药品检验所,贵阳550004;贵州省食品药品检验所,贵阳550004【正文语种】中文【中图分类】R965【相关文献】1.HPLC同时测定紫朱软膏中左旋紫草素和β,β'-二甲基丙烯酰阿卡宁含量 [J], 丁叶芳;李西林;柳国斌;张钰泉2.薄层扫描法测定新疆不同产地新疆紫草中β,β'-二甲基丙烯酰阿卡宁的含量 [J], 徐海燕;丁文欢;张慧;王延蛟3.紫草中β,β'-二甲基丙烯酰阿卡宁提取和含量测定及其稳定性研究 [J], 谢清春;陈燕忠;吕竹芬4.基于新疆紫草中β,β'-二甲基丙烯酰阿卡宁含量变化的浓缩工艺研究 [J], 王佩;汪斌;刘康;陈俊;葛海涛5.不同产地蒙紫草中左旋紫草素和β,β'-二甲基丙烯酰阿卡宁含量测定及质量标准研究 [J], 吕晓洁;渠弼;刘德旺;程丹丹;那生桑因版权原因,仅展示原文概要,查看原文内容请购买。

紫外可见分光光度法测定呼和-嘎日迪-13散中紫草的含量成志平
【期刊名称】《中国民族医药杂志》
【年(卷),期】2016(022)009
【摘要】目的:紫外可见分光光度法测定呼和-嘎日迪-13散中紫草的含量.方法:采用紫外可见分光光度法,样品以石油醚(60~90℃)超声振荡提取.结果:左旋紫草素在8.264~28.924μg/mL范围内呈良好的线性关系,r=0.99982,平均回收率
96.2%,RSD为0.43%,结论:本法灵敏、准确,操作简单,重复性好.
【总页数】2页(P55-56)
【作者】成志平
【作者单位】包头市食品药品检验检测中心,内蒙古包头014060
【正文语种】中文
【中图分类】R291.2
【相关文献】
1.HPLC法测定蒙药嘎日迪-13味丸中甘草苷、甘草酸铵、木香烃内酯、去氢木香内酯含量
2.嘎日迪-13和嘎日迪-15中14种元素含量的比较研究
3.HPLC法测定蒙药哈敦嘎日迪-13丸中总没食子酸的含量
4.显微定量法测定蒙成药嘎日迪─13中珍珠的含量
5.HPLC法测定蒙药哈敦嘎日迪-13丸中木香烃内酯和去氢木香内酯的含量
因版权原因,仅展示原文概要,查看原文内容请购买。


HPLC 法测定大鼠血浆及粪便中新疆紫草3种萘醌类色素的含量古力扎努·阿合买提;买尔旦·马合木提【期刊名称】《新疆医科大学学报》【年(卷),期】2016(39)8【摘要】目的:建立测定大鼠血浆及粪便中新疆紫草3种萘醌类成分含量的高效液相色谱法(HPLC 法)。
方法采用 COSMOIL C18-AR-Ⅱ(4.6 mm×250 mm,5μm)色谱柱,流动相 A 乙腈(A)-0.15%甲酸(B)(v/v)溶液,进行洗脱,流速1.0 mL/min,检测波长275 nm ,柱温25℃,检测时间35 min。
结果血浆中左旋紫草素在0.40~6.40μg、乙酰紫草素在25.0~600.0μg、β,β′-二甲基丙烯酰紫草素在25.0~600.0μg 范围内均与峰面积呈线性关系,左旋紫草素回收率为97.78%~100.1%、乙酰紫草素为100.1%~100.2%、β-β′-二甲基丙烯酰紫草素为97.78%~100.1%,日间、日内 RSD 均<1.5%。
粪便中各组分在10~400μg/mL 范围内与峰面积呈线性关系,平均回收率分别为(78.2±2.5)%、(82.0±2.9)%和(79.5±3.5)%。
0~12、12~24、24~48 h 粪便中左旋紫草素绝对回收量分别为23.61、38.28、4.13 mg;乙酰紫草素分别为7.20、70.78、11.53 mg;β-β′二甲基丙烯酰紫草素分别为46.32、60.15、16.88 mg。
结论HPLC 法准确可靠,重复性好,适用于测定血浆及粪便中新疆紫草萘醌类成分含量。
%Objective To establish a HPLC method for determine of three contents of naphthaquinone from Arnebia euchroma (Royle)Johnst in rats plasma and faeces.Methods COSMOIL C18-AR-Ⅱ (4.6 mm× 250 mm,5μm),column was used with acetonitrile (A)-0.15% formic acid (B)(v/v)as mobile phase by gradient eluting with the flow rate of 1.0 mL/min for 35 min,and the colum n temperature was maintained at 25℃,detecting wavelength at 274 nm.Results The linear range in plasma ofshikonin,acetylshikonin andβ-β,-dimethylacrylshikonin were between 0.40 to 6.40 μg,25.0 to 600.0 μg and 25.0 to 600.0 μg,re-spectively.The recoveries of these ingredients correspondingly were between 97.78 to 100.1%,100.1 to100.2% and 97.78 to100.1%.The RSD of intra-day and inter-day were less than 1.5%.The linear range in faeces ofshikonin,acetylshikonin andβ-β,-dimethylacrylshikonin were between 10-400 μg/mL with the average recoveries of (78.2 ± 2.5)%,(82.0 ± 2.9)% and (79.5 ± 3.5)%,respectively.Absolute recovery amount of Naphthoquinone in feces as:shikonin 0-12 h:23.61 mg,12-24 h:38.28 mg,24-48 h:4.13mg;acetylshikonin 0-12 h:7.20 mg,12-24 h:70.78 mg,24-48 h:11.53 mg andβ-β,-dimeth-ylacrylshikonin 0-12 h:46.32 mg,12-24 h:60.15 mg,24-48h:16.88 mg.Conclusion The established method has a good selectivity and reproducibility for the determination of naphthaquinone from Arnebia euchroma (Royle)Johnst in plasma and faeces of rats.【总页数】5页(P1012-1016)【作者】古力扎努·阿合买提;买尔旦·马合木提【作者单位】新疆医科大学 1 药学院;附属肿瘤医院,乌鲁木齐 830011【正文语种】中文【中图分类】R914【相关文献】1.HPLC法测定紫草中萘醌类成分 [J], 曾祖平;韩旭阳;王宏;彭冰;石佳2.紫外-可见分光光度法测定紫草油中羟基萘醌总色素的含量 [J], 苗颖;李英男;李栋;梁莹莹;张淑君;陆进;赫军;马秉智;杨丽3.正交试验优选新疆紫草中羟基萘醌类色素的提取工艺研究 [J], 永春;锡林通拉嘎;4.HPLC法测定新疆软紫草属4种植物根中萘醌类成分 [J], 丁文欢;燕雪花;葛亮;徐海燕5.HPLC法同时测定泰山紫草中5种萘醌类成分的含量 [J], 董娜;高召军;曲晓兰;齐永秀因版权原因,仅展示原文概要,查看原文内容请购买。

UV法测定紫草中羟基萘醌总色素含量的不确定度评定及应用作者:关卓明何敬铭林泳仪李砺来源:《品牌与标准化》2024年第01期【摘要】对采用紫外-可见分光光度(UV)法测定紫草中羟基萘醌总色素含量全过程进行分析,建立数学模型,确定影响总色素测定不确定度的因素并对其进行评估。
结果表明,总色素含量测定的扩展不确定度为0.06%,测定结果表示为(1.69±0.06)%(k=2),能力验证结果为满意。
【关键词】紫草;羟基萘醌;分光光度法;能力验证;不确定度【DOI编码】10.3969/j.issn.1674-4977.2024.01.009Determination of Total Hydroxynaphthoquinone Pigment Content in Purple Grass by Spectrophotometry Evaluation of Uncertainty and Application in Capability VerificationGUAN Zhuoming, HE Jingming, LIN Yongyi, LI Li*(Sinopharm group fengliaoxing 〔Foshan〕herbal medicine decoction pieces Co., Ltd., Foshan 528000, China)Abstract: To verify the ability of UV spectrophotometry to determine the total content of hydroxynaphthoquinone pigments in purple grass, establish a mathematical model, determine the factors that affect uncertainty, and evaluate each factor. Results showed that the expanded uncertainty of the total pigment content determination was 0.06%, and the determination result was expressed as (1.69±0.06)% (k=2), indicating a satisfactory capability validation result.Keywords: hydroxyanaphthoquinone; spectrophotometry; capability verification;uncertainty紫草的使用历史非常悠久。

紫草的提取实验报告
紫草提取实验报告
摘要:
本实验旨在探究紫草的提取过程,并对提取物进行分析。
实验结果表明,采用乙醇提取法可以获得较高的提取率,并且提取物中含有丰富的活性成分。
这为进一步研究紫草的药用价值提供了重要的参考。
引言:
紫草是一种常见的中草药材,具有清热解毒、活血化瘀的功效,被广泛应用于中医药领域。
为了充分发挥紫草的药用价值,提取紫草中的有效成分是非常重要的。
因此,本实验旨在探究紫草的提取过程,并对提取物进行分析,为进一步研究紫草的药用价值提供参考。
材料与方法:
1. 实验材料:紫草干燥植物材料、乙醇、蒸馏水
2. 提取方法:将紫草粉末与乙醇按一定比例混合,加热回流提取,得到提取液后蒸馏除去乙醇,得到提取物
3. 提取物分析:使用高效液相色谱法对提取物中的活性成分进行分析
结果与讨论:
经过实验,我们发现采用乙醇提取法可以获得较高的提取率,且提取物中含有丰富的活性成分。
这说明乙醇提取法是一种有效的提取紫草有效成分的方法。
此外,通过高效液相色谱法分析,我们发现提取物中含有多种活性成分,如紫草素、紫草酸等。
这些活性成分是紫草药用价值的重要组成部分,对于紫草的药用研究具有重要意义。
结论:
本实验结果表明,采用乙醇提取法可以获得较高的提取率,并且提取物中含有丰富的活性成分。
这为进一步研究紫草的药用价值提供了重要的参考。
我们相信,通过对紫草提取物的进一步研究,可以更好地发挥紫草的药用价值,为人类健康事业做出贡献。
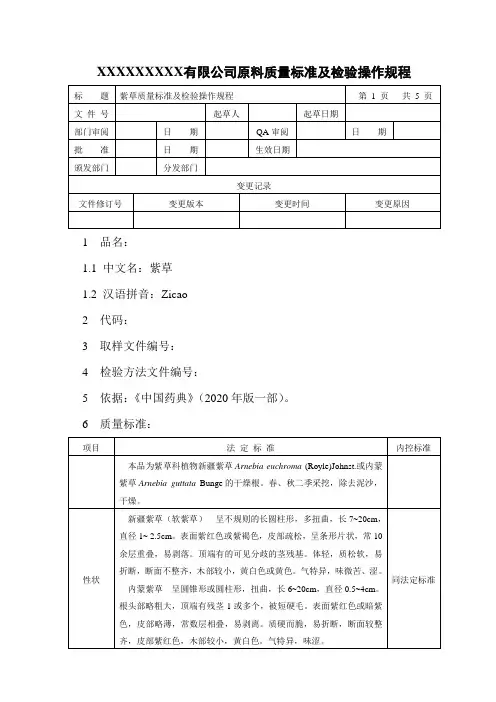
XXXXXXXXX有限公司原料质量标准及检验操作规程1 品名:1.1 中文名:紫草1.2 汉语拼音:Zicao2 代码:3 取样文件编号:4 检验方法文件编号:5 依据:《中国药典》(2020年版一部)。
6 质量标准:7 检验操作规程:7.1 试药与试剂:石油醚(60~90℃)、紫草对照药材、环己烷、甲苯、乙酸乙酯、甲酸、氢氧化钾、甲醇、乙醇、乙腈、β,β′-二甲基丙烯酰阿卡宁对照品。
7.2 仪器与用具:电子天平、显微镜、烘箱、硅胶G薄层板、展开缸、高效液相色谱仪、二氧化硫测定仪、紫外分光光度计。
7.3 性状:取本品适量,自然光下目测色泽,嗅闻气味。
7.4 鉴别:7.4.1 取本品制片置10×10显微镜下做显微观察。
8.4.2 取本品粉末0.5g,加石油醚(60~90℃)20ml,超声处理20分钟,滤过,滤液浓缩至lml,作为供试品溶液。
另取紫草对照药材0.5g,同法制成对照药材溶液。
照薄层色谱法(附录7)试验,吸取两种溶液各4μl,分别点于同一硅胶G薄层板上,以环己烷-甲苯-乙酸乙酯-甲酸(5:5:0.5:0.1)为展开剂,展开,取出,晾干。
供试品色谱中,在与对照药材色谱相应的位置上,显相同的紫红色斑点;再喷以10%氢氧化钾甲醇溶液,斑点变为蓝色。
7.5 检查7.5.1水分不得过15.0%(附录15 第二法)。
7.5.2二氧化硫残留量照二氧化硫残留量测定法(附录58)测定,不得过150mg/kg。
7.6 含量测定:羟基萘醌总色素取本品适量,在50℃干燥3小时,粉碎(过三号筛),取约0.5g,精密称定,置100ml量瓶中,加乙醇至刻度,4小时内时时振摇,滤过。
精密量取续滤液5ml,置25ml量瓶中,加乙醇至刻度,摇匀。
照紫外-可见分光光度法(附录4),在516nm波长处测定吸光度,按左旋紫草素)为242计算,即得。
(C16H16O5)的吸收系数系数为(E1%1cm本品含羟基萘醌总色素以左旋紫草素(C16H16O5)计,不得少于0.80%。

复方紫草油的制备工艺与质量标准研究目的:验证复方紫草油的制备工艺,提高质量标准控制水平。
方法:以左旋紫草素的含量为指标,考察制备工艺的合理性。
采用薄层色谱法对白芷、忍冬藤、冰片进行鉴别,用高效液相色谱法测定左旋紫草素的含量。
结果:制备工艺过程中左旋紫草素稳定,原制备工艺合理;白芷、忍冬藤、冰片的鉴别斑点清晰,阴性无干扰;左旋紫草素测定方法经方法学验证,结果可行。
结论:结果表明,制备工艺合理,新建立的质量标准可行。
[Abstract] Objective: To study the preparation technology of Compound Radix Arnebiae Oil and its feasibility, and to improve the quality standard level. Methods: The preparation technology was studied by determining the stability of L-shikonin during the time of preparation. Angelica dahurica, Honeysuckle stem, Borneol in Compound Radix Arnebiae Oil were identified by TLC. The content of L-shikonin was determined by HPLC. Results: L-shikonin was stable during the time of preparation. The preparation technology was feasible. The dots of TLC were clear, separation degree was good, and specific attribute was strong. The negative control showed no interference.The HPLC method was able to determine the content of L-shikonin. Conclusion: Preparation technology is feasible, and the quality of Compound Radix Arnebiae Oil can be controlled.[Key words] Compound Radix Arnebiae Oil;Preparation technology;Quality standard; L-shikonin复方紫草油(烧伤油)系我院自制制剂,由紫草、忍冬藤、冰片、白芷、石蜡、香油等药材煎制而成,临床应用20多年,本品对各种烧伤疗效果确切。
高效液相色谱法测定紫草油中β,β'-二甲基丙烯酰阿卡宁的含量王贵英;骆勇;左宏笛【摘要】目的:建立紫草油中β,β'-二甲基丙烯酰阿卡宁的含量测定方法.方法:采用高效液相色谱(HPLC)法,色谱柱为迪马C18钻石色谱柱(250 mm×4.6mm,5μm);流动相为乙腈-水-甲酸(75∶25∶0.2);检测波长为275 nm;柱温为室温.结果:在80.6 ~564.5 ng范围内,β,β'-二甲基丙烯酰阿卡宁进样量和峰面积具有良好的线性关系,线性回归方程为C=1.027 9×10-6A +2.358 9×10-2,r =0.999 9.测得紫草油中β,β'-二甲基丙烯酰阿卡宁平均含量为0.869 2 mg/ml.结论:该方法简便可行,重现性好,可作为紫草油的质量控制方法.【期刊名称】《中国医院用药评价与分析》【年(卷),期】2013(013)012【总页数】3页(P1089-1091)【关键词】紫草油;高效液相色谱法;β,β'-二甲基丙烯酰阿卡宁;含量测定【作者】王贵英;骆勇;左宏笛【作者单位】贵州省食品药品检验所,贵阳550004;贵州省食品药品检验所,贵阳550004;贵州省食品药品检验所,贵阳550004【正文语种】中文【中图分类】R965紫草油是贵阳钢厂职工医院的院内制剂,单以紫草为主药、菜油为溶剂制成,是供外用的油状液体制剂,具有凉血、活血、解毒透疹之功,临床上用于治疗湿疹、烧伤、烫伤、小儿尿布疹。
经多年的临床实践,疗效确切。
由于现行医疗机构制剂规范中紫草油的质量标准较为简单,无含量测定项。
现参照《中华人民共和国药典》中紫草的质量控制方法,采用高效液相色谱(HPLC)法测定紫草油中β,β'-二甲基丙烯酰阿卡宁的含量,结果如下。
1 材料1.1 仪器LC-20A HPLC仪(日本岛津公司生产);LCsolution色谱工作站。
龙源期刊网 http://www.qikan.com.cn 紫朱软膏中紫草的质量控制 作者:丁叶芳 李西林 柳国斌 牛东霞 来源:《中国中医药信息》2015年第01期
摘要:目的 建立紫朱软膏中紫草的质量控制方法。方法 采用薄层色谱法对紫朱软膏中紫草进行定性鉴别。以左旋紫草素含量为评价指标,采用正交试验设计优选左旋紫草素的提取工艺,以紫外可见分光光度法对左旋紫草素进行含量测定。结果 在薄层色谱中,主斑点清晰,阴性对照无干扰。左旋紫草素的最佳提取工艺为:提取3次,每次30 min,NaOH体积分数为1%、用量为50 mL。左旋紫草素在0.010 2~0.035 7 mg(r2=0.999 7)范围内与吸收度呈良好的线性关系,平均加样回收率为103.27%,RSD=4.17%。结论 本方法简便可行,可用于紫朱软膏的质量控制和评价。
关键词:紫朱软膏;紫草;左旋紫草素;质量控制 中图分类号:R284.1 文献标识码:A 文章编号:1005-5304(2015)01-0077-04 紫朱软膏是上海中医药大学附属曙光医院用于治疗糖尿病足的经验方,由紫草、朱砂、冰片、黄芪、龙血竭、阿胶6味药组成,具有清热解毒、祛腐生肌、补气益血功效。紫草作为处方君药,具有抑菌消炎作用,对金黄色葡萄球菌、绿脓杆菌等有明显抑制作用,并能促进上皮生长,加速创口愈合[1-2]。紫草含多种活性成分,其中主要是以左旋紫草素为主的羟基萘醌类色素,以左旋紫草素为指标性成分对羟基萘醌总色素进行质量控制是保证紫朱软膏发挥临床疗效的必要内容。本研究采用薄层色谱法对紫朱软膏中的紫草进行定性鉴别,运用正交试验法优选左旋紫草素的提取工艺,并采用紫外可见分光光度法对紫朱软膏中羟基萘醌总色素进行含量测定,建立该制剂的质量标准。
1 仪器与试药 Agilent 8453型紫外分光光度计(美国Agilent公司),Sartorius BSA323S电子天平、Sartorius BT125D电子天平(北京赛多利斯科学仪器有限公司),BRANSON B550S-MT超声清洗仪(必能信超声上海有限公司),Centrifuge 54300R型冷冻离心机(德国eppendorf公司),旋转蒸发仪(BUCHI公司)。
HPLC 法测定大鼠血浆及粪便中新疆紫草3种萘醌类色素的含量古力扎努·阿合买提;买尔旦·马合木提【摘要】目的:建立测定大鼠血浆及粪便中新疆紫草3种萘醌类成分含量的高效液相色谱法(HPLC 法)。
方法采用 COSMOIL C18-AR-Ⅱ(4.6 mm×250 mm,5μm)色谱柱,流动相 A 乙腈(A)-0.15%甲酸(B)(v/v)溶液,进行洗脱,流速1.0 mL/min,检测波长275 nm ,柱温25℃,检测时间35 min。
结果血浆中左旋紫草素在0.40~6.40μg、乙酰紫草素在25.0~600.0μg、β,β′-二甲基丙烯酰紫草素在25.0~600.0μg 范围内均与峰面积呈线性关系,左旋紫草素回收率为97.78%~100.1%、乙酰紫草素为100.1%~100.2%、β-β′-二甲基丙烯酰紫草素为97.78%~100.1%,日间、日内 RSD 均<1.5%。
粪便中各组分在10~400μg/mL 范围内与峰面积呈线性关系,平均回收率分别为(78.2±2.5)%、(82.0±2.9)%和(79.5±3.5)%。
0~12、12~24、24~48 h 粪便中左旋紫草素绝对回收量分别为23.61、38.28、4.13 mg;乙酰紫草素分别为7.20、70.78、11.53 mg;β-β′二甲基丙烯酰紫草素分别为46.32、60.15、16.88 mg。
结论HPLC 法准确可靠,重复性好,适用于测定血浆及粪便中新疆紫草萘醌类成分含量。
%Objective To establish a HPLC method for determine of three contents of naphthaquinone from Arnebia euchroma (Royle)Johnst in rats plasma and faeces.Methods COSMOIL C18-AR-Ⅱ (4.6 mm× 250 mm,5μm),column was used with acetonitrile (A)-0.15% formic acid (B)(v/v)as mobile phase by gradient eluting with the flow rate of 1.0 mL/min for 35 min,and the colum n temperature was maintained at 25℃,detectingwavelength at 274 nm.Results The linear range in plasma ofshikonin,acetylshikonin andβ-β,-dimethylacrylshikonin were between 0.40 to 6.40 μg,25.0 to 600.0 μg and 25.0 to 600.0 μg,re-spectively.The recoveries of these ingredients correspondingly were between 97.78 to 100.1%,100.1 to100.2% and 97.78 to100.1%.The RSD of intra-day and inter-day were less than 1.5%.The linear range in faeces ofshikonin,acetylshikonin andβ-β,-dimethylacrylshikonin were between 10-400 μg/mL with the average recoveries of (78.2 ± 2.5)%,(82.0 ± 2.9)% and (79.5 ± 3.5)%,respectively.Absolute recovery amount of Naphthoquinone in feces as:shikonin 0-12 h:23.61 mg,12-24 h:38.28 mg,24-48 h:4.13mg;acetylshikonin 0-12 h:7.20 mg,12-24 h:70.78 mg,24-48 h:11.53 mg andβ-β,-dimeth-ylacrylshikonin 0-12 h:46.32 mg,12-24 h:60.15 mg,24-48h:16.88 mg.Conclusion The established method has a good selectivity and reproducibility for the determination of naphthaquinone from Arnebia euchroma (Royle)Johnst in plasma and faeces of rats.【期刊名称】《新疆医科大学学报》【年(卷),期】2016(039)008【总页数】5页(P1012-1016)【关键词】新疆紫草;左旋紫草素;乙酰紫草素;β,β′-二甲基丙烯酰紫草素;HPLC;大鼠血浆;大鼠粪便【作者】古力扎努·阿合买提;买尔旦·马合木提【作者单位】新疆医科大学 1 药学院;附属肿瘤医院,乌鲁木齐 830011【正文语种】中文【中图分类】R914新疆紫草[Arnebia euchroma (Royle) Johnst]习称软紫草,又名假紫草、新疆软紫草、新疆假紫草, 为紫草科多年生草本植物新疆紫草的根, 分布于我国新疆、甘肃及西藏西部地区, 是药材紫草的主要来源[1]。
HPLC法测定紫草中的左旋紫草素的含量
摘要:目的:建立紫草中左旋紫草素的含量测定方法。
方法:采用HPLC 法,色谱柱为YWG-C-18,流动相为甲醇-0.025mol/L磷酸(80∶20),测定波长:516nm,柱温为25℃,流速为1.0mL/min,进样量为20μL,结果:左旋紫草素在0.176~0.880μg间,左旋紫草素进样量与峰面积呈良好的线性y=2×107x+31103,R2=0.9993,测得紫草中左旋紫草素的平均含量为1.78%。
结论:应用HPLC法测定紫草中的左旋紫草素含量,结果准确,重现性好。
关键词:紫草;左旋紫草素;HPLC;含量测定
紫草(Alkanna Tinctoria)是传统中药之一,属于紫草科多年生草本植物,以其干燥根入药。
《中国药典》有收载,谓其性寒、味甘咸、归心肝经,具有凉血、活血、解毒透疹、补中益气作用。
紫草的药理作用很多,如:抗生育作用、抗菌、抗炎、抗癌、降糖、保肝及提高肌体免疫功能。
紫草中的有效成分为紫草宁衍生物,属萘醌类色素[1]。
《中国药典》2005版采用紫外分光光度法测定左旋紫草素的含量。
本实验采用HPLC(高效液相色谱)法对紫草中的左旋紫草素进行含量检测。
1 仪器与试剂
实验中紫草由亚洲大药房提供,左旋紫草素对照品由中国生物制品药品检定所提供,其余试剂均为分析纯。
高效液相色谱仪(紫外可见检测器,LC-05Pi液体泵),AG204电子天平,KQ2200B超声机
2 实验方法与结果
2.1 色谱条件[2,3]
色谱柱:YWG-C-18柱;流动相:甲醇-0.025mol/l磷酸(80∶20);柱温:25℃;流速:1.0mL/min;测定波长:516nm。
按左旋紫草素计算理论塔板数为2500(>103)。
在此条件下,样品中左旋紫草素与其他相关峰均能达到基本分离。
2.2 对照品溶液的配制
精密称定5.5mg左旋紫草素对照品置10mL容量瓶中,用甲醇定容到10mL,得0.55mg/mL对照品溶液。
用移液管精密移取4mL于50mL容量瓶中,用甲醇稀释定容到50mL,得浓度为0.044mg/mL对照品溶液。
2.3 供试品溶液的配制[4]
称取紫草10g,加入95%乙醇100mL,超声20min。
放冷过滤,滤渣加入95%乙醇50mL超声10min。
放冷过滤,合并滤液,加入2%的氢氧化钠溶液使溶液
呈碱性(变蓝色),过滤,滤液加酸调pH到呈酸性(变红色),过滤,滤液减压浓缩回流至20mL左右。
在50℃下进行干燥。
精密称定提取物的重量,把提取物加入甲醇,超声溶解,定容在100mL的容量瓶中。
2.4 线性关系与标准曲线[5]
精密移取已经配制好的0.044mg/mL标准品溶液 2.0mL,4.0mL,6.0mL,8.0mL,10.0mL于10mL容量瓶中,加甲醇定容至刻度,分别吸取20ul,注入液相色谱中,测定色谱峰面积,得回归方程y=2×107x+31103,R2=0.9993。
2.5 精密度考察
用上述方法制备的供试品连续进6针,测定峰面积。
考察相对标准偏差。
见下表:
结果:精密度考察符合要求。
2.6 回收率的测定[6]
方法:取紫草粉末两份,各10g精密称定,置100mL具塞三角瓶中,一份加入左旋紫草素对照品溶液(0.044mg/mL)20mL作为对照品溶液;另一份不加作为样品溶液。
然后各加入95%乙醇100mL超声20min。
放冷过滤,滤渣加入95%乙醇50mL超声10min。
放冷过滤,合并滤液,减压浓缩回流至20mL左右。
在50℃下进行干燥。
精密称定提取物的重量,把提取物加入甲醇超声溶解,定容在100mL的容量瓶中。
见表2:
结果:回收率符合要求。
2.7 紫草中左旋紫草素的含量测定
方法:将不同批次的中药紫草制备成供试品进针,测定含量。
见表3:
3 讨论
(1)在紫草的提取过程中,浸出液要浓缩到溶液体积20mL左右,使得乙醇的含量很少,才能利用加碱使紫草素溶于水相,过滤后加酸使沉淀析出。
在加碱时不要剧烈摇动、搅拌,以免产生乳化效果,不利于提取;加酸时要使溶液变成红色,静置析出。
(2)由于紫草中紫草素含量很低,所以在进液相时浓度要比较大,使得误差能够减少。
进针后,其色谱峰周期过长,在对照品出峰时间过后可以将流速由1.0mL/min提高到1.5mL/min。
(3)测定方法的选择。
有文献报道用紫外分光光度法及薄层扫描测定紫草素的含量。
本实验采用高效液相色谱法测定紫草中的左旋紫草素的含量,结果准确、重现性好、灵敏度高。
(4)样品测定结果表明,通过对样品颜色的观察发现,左旋紫草素含量高的成品颜色深红,而所测含量较低的成品颜色较浅,这可能与紫草原药材的质量有关。
参考文献:
[1] 卢贺起,丁家欣,魏雅川.紫草的实验研究进展[J].中国中医药信息,1998,5(1):20.
[2] 马瑛,王嗣芩,黄猛.HPLC测定紫草中紫草素含量[J].中国药学,2000,35(20):98.
[3] 李忠良,张敏,郭兰建.紫草中紫草素的含量测定[J].药物分析,1986,6(1):41.
[4] 杨晓笛,卢锋,黄美充.中药紫草有效成分的提取[J].汕头大学学报,2002,17(4):67.
[5] 孟宪生,沙明,曹爱民,张东方.HPLC法测定紫草及其制剂紫金油中左旋紫草素的含量[J].中草药,2003,34(7):615.
[6] 陈发奎.常用中草药有效成分含量测定[M].北京:人民卫生出版社,1997:732~732.。