不同柱温模式毛细管柱气相色谱法测试VFA结果的比较
- 格式:pdf
- 大小:769.46 KB
- 文档页数:3

毛细管柱是我们平时做气相色谱分析必不可少的重要耗材,毛细管柱分离性能的好坏直接影响我们实验结果的合格率。
经常使用毛细管柱的实验小伙伴对于毛细管柱的型号应该都不陌生吧,例如一款色谱柱CD-5MS,30m*0.25mm*0.25um,从这些描述中我们可以获得色谱柱的固定相类型,长度,内径和膜厚。
今天小编给您分享毛细管柱的一些知识,希望对您的实验有所帮助。
01 我们常用的毛细管柱的型号有哪些呢?一般从色谱柱的极性由弱到强我们常用的色谱柱包括-1、-5、-35、-50、-624、-1701、-WAX,他们的固定相分别是100%甲基聚硅氧烷、5%苯基-95%甲基聚硅氧烷、35%苯基-65%甲基聚硅氧烷、50%苯基-50%甲基聚硅氧烷、6%氰丙基苯基-94%甲基聚硅氧烷、14%氰丙基苯基-86%甲基聚硅氧烷、聚乙二醇20M。
当然现在也有越来越多的专用柱被大家所熟知,例如脂肪酸甲酯专用柱广泛用来作为食品中脂肪酸的测试分析;血液中酒精检测专用柱用来作为人的血液酒精分析等。
另外还有-1MS、-5MS、-1HT、-5HT的色谱柱,这些后端有MS、HT 后缀的柱子,MS柱主要是流失更低适用于质谱分析,能够尽量降低离子源的污染,减少质谱维护工作、HT柱温度耐受更强,适用于检测例如棕榈酸等需要较高分离温度的化合物检测。
02 针对这么多的色谱柱种类,如何去判断和验证这些色谱柱的性质呢?那就需要了解色谱柱型性能评价的理论知识。
主要包括色谱柱的分离能力、极性以及色谱柱的柱活性。
一般从这三个方面入手就能对色谱柱有充分的了解了。
色谱柱的分离能力主要包括塔板数、总分离效能、分离数和涂渍效率。
塔板数大家都不陌生,是评价柱效的主要指标,有有效塔板数和理论塔板数之分。
总分离效能是指色谱柱在一定条件下对混合物的分离能力。
一般以分离度来判断。
分离数是指在相邻同系物峰之间可插入的组分峰的数目,也是指色谱柱的分离能力,是总分离效能的部分体现。
涂渍效率和生产工艺有关,是表征空柱柱效达到最理想化的程度,一般非极性柱可以到90%以上,极性柱只能在60%-70%左右。

一、固定液使用固定相: AT SE-30,AT OV-1组成100%甲基聚硅氧烷极性非极性应用碳氢化合物同类型号DB(HP) -1、AC1、SPB-1、CPSIL5、DM-1、RT-1 使用温度50—300℃固定相: AT OV-101组成100%甲基聚硅氧烷(胶体)极性非极性应用氨基酸、基油同类型号HP-101、AC1 、SP-2100使用温度0—350℃固定相: AT SE-52AT SE-54组成5%苯基甲基聚硅氧烷,1%乙烯基5%苯基甲基聚硅氧烷极性非极性应用多核芳烃、酚、酯、碳氢化合物、药物、醇同类型号DB(HP)-5、AC5、SPB-5、DM-5、CPSiL8、Rtx-5 使用温度50—350℃固定相: AT OV-1701组成7%氰丙基7%苯基甲基聚硅氧烷极性非极性应用药物、醇、酯、硝基化合物同类型号DB(HP)-1701、AC10、DB-1701、SPB-1701、RT-1701、CP-Sil 19CB 使用温度0—280℃固定相: AT XE-60组成25%氰乙基甲基聚硅氧烷极性中极性应用酯、硝基化合物同类型号DB (HP) -225、AC225使用温度0—280℃固定相: AT OV-17组成50%苯基甲基聚硅氧烷极性中极性应用药物、农药同类型号DB(HP)-17、AC10使用温度0—250℃固定相: AT FFAP组成聚乙二醇—TPA改性极性极性应用酸、醇、醛、酯、腈、酮、基油同类型号DB (HP) –FFAP、SP-1000、Supecl-NUKOL、AC20使用温度50—250℃固定相: AT PEG-20M组成聚乙二醇—20M极性极性应用同类型号HP- Wax、DB-Wax、AC20使用温度50—200℃固定相: AT 农残Ⅰ号AT 农残Ⅱ号组成极性应用六六六、DDT等八种含氯农药拟除虫菊酯类、含磷类农药同类型号SPB-608、HP-608使用温度25—300℃二、毛细管柱内径0.53mm 具有近似填充柱的负荷量,总柱效则远远超过填充柱。


挥发性脂肪酸的测定一、GC(Gas chromatograph)工作条件仪器:GC-14B型气相色谱仪(日本岛津公司)毛细柱:NUKOLTM Capillary Column ( Supelco );Column No.34292-07B30m×0.32mm×0.25μm film thickness气相色谱仪参数:色谱柱采用毛细吸管柱,柱温130℃,汽化温度180℃,采用氢离子火焰检测器,检测温度180℃,载气为氮气,压力为60 KPa,氢气压力为50 KPa,氧气压力50 KPa,灵敏度(档)为101,衰减 3.0二、样品制备1.试剂制备(1)配制25%w/v 的偏磷酸溶液,将25克偏磷酸溶在100mL双蒸水中(2)巴豆酸的配制,在100ml的偏磷酸溶液中加入0.6464g的巴豆酸,定容到100mL。
(3)标准样品,准确称取色谱标准级乙酸0.9100 g、丙酸0.3700 g、丁酸0.1765 g、异丁酸0.1765g、戊酸和异戊酸分别为0.1985g,分别溶于双蒸水中,再各自定容至100 mL。
2.样品制备(1) 发酵液取(或过滤瘤胃液)VFA测定1ml样品到离心管中,再加入0.2 ml的偏磷酸巴豆酸混合溶液,-20℃冰箱保存过夜,解冻后12000rpm离心10min,取上清液保存,测定前再12000rpm 离心10min(或少许通过0.22μm针式滤器,滤液直接进样用 1.0微升微量进样器瞬时注入色谱仪,进样量为0.2-1.0μL。
(2) 食糜及粪样VFA测定取1克置于离心管中,加入5-10倍的双蒸水,混合均匀。
取1mL上清夜,加偏磷酸巴豆酸0.2mL/mL。
其它步骤同上。
3.保留时间的确定与样品处理相同,在 1 ml标准样品中加入0.2mL偏磷酸与巴豆酸的混合样,上样测出乙酸、丙酸、丁酸、异丁酸、戊酸和异戊酸的保留时间。
4.有机酸浓度的计算通过标准样品和内标巴豆酸各自的(或浓度)和峰面积可以计算出乙酸、丙酸及丁酸等有机酸的相对校正因子,然后根据乙酸、丙酸及丁酸的重量(或浓度)与其峰面积成正比计算出各个样品中的乙酸、丙酸及丁酸浓度。


两种分析聚合级乙烯中烃类杂质的气相色谱方法对比刘建英两种分析聚合级乙烯中烃类杂质的气相色谱方法对比【摘要】通过对PLOT Al2O3“M”石英毛细管柱和填充柱分离方法的比较,采用优化的气相色谱条件,来分析聚合级乙烯中的烃类杂质。
结果表明用PLOT Al2O3“M”石英毛细管柱采用程序升温法分离,分流进样,用氢火焰离子化检测器检测,外标法定量,灵敏度高,对不同浓度的样品进行重复性测试的相对标准偏差为%,准确度及精密度均符合要求,并且操作简单,比原方法缩短了分析时间。
【关键词】气固色谱法氢火焰离子化检测器外标法聚合级乙烯1 前言随着信息时代的发展,我国的各行各业都在突飞猛进,化学工业更是飞速发展,齐鲁公司聚乙烯生产已突破60万吨/年,对于乙烯的质量要求更加严格。
乙烯作为塑料企业的主要生产原料,其质量直接影响到装置的稳定生产,其中痕量杂质,如乙炔对聚烯烃产品的线性度、重均分子量MW分布都会产生不利的影响。
全国实行ISO9002质量体系认证,为了达到这一质量要求,就必须对以前的分析方法进行更新,严格控制乙烯的纯度及烃类杂质,使我们生产的产品满足用户的质量要求。
过去分析聚合级乙烯中的烃类杂质采用两根填充柱(20%磷酸三丁酯柱,柱长9米;硅胶柱,柱长4米),两次进样分析来达到烷烃、烯烃、炔烃的分离。
以上方法存在操作繁琐,分离效果差,影响准确定量。
经过一系列分析对比采用PLOT Al2O3 “M”石英毛细管柱,分流进样系统后,进样量为1ml(原来为ml ),柱长比原来增大5~10倍,理论塔板数为26287块,完全符合Q/—2006《中国石化齐鲁股份有限公司塑料厂企业标准—聚合级乙烯》要求,烃类杂质能完全分离,检测限可达%, 分析速度快,准确度、精密度高。
2 设备和材料材料及试剂五组分混合标准气,组成见表1(大连气体开发公司生产)。
表1 标准样品及各组分浓度仪器惠普HP5890型气相色谱仪,氢火焰检测器,HP3396型数据处理机,六通阀进样,1ml定量管。

挥发性脂肪酸的测定一、GC(Gas chromatograph)工作条件仪器: GC-14B型气相色谱仪(日本岛津公司)毛细柱:NUKOLTM Capillary Column ( Supelco );Column No.34292-07B30m×0.32mm×0.25μm film thickness气相色谱仪参数:色谱柱采用毛细吸管柱,柱温130℃,汽化温度180℃,采用氢离子火焰检测器,检测温度180℃,载气为氮气,压力为60 KPa,氢气压力为50 KPa,氧气压力50 KPa,灵敏度(档)为101,衰减 3.0二、样品制备1.试剂制备(1)配制25%w/v 的偏磷酸溶液,将25克偏磷酸溶在100mL双蒸水中(2)巴豆酸的配制,在100ml的偏磷酸溶液中加入0.6464g的巴豆酸,定容到100mL。
(3)标准样品,准确称取色谱标准级乙酸0.9100 g、丙酸0.3700 g、丁酸0.1765 g、异丁酸0.1765g、戊酸和异戊酸分别为0.1985g,分别溶于双蒸水中,再各自定容至100 mL。
2.样品制备(1) 发酵液取(或过滤瘤胃液)VFA测定 1ml样品到离心管中,再加入0.2 ml的偏磷酸巴豆酸混合溶液,-20℃冰箱保存过夜,解冻后12000rpm离心10min,取上清液保存,测定前再12000rpm 离心10min(或少许通过0.22μm针式滤器,滤液直接进样用 1.0微升微量进样器瞬时注入色谱仪,进样量为0.2-1.0μL。
(2) 食糜及粪样VFA测定取1克置于离心管中,加入5-10倍的双蒸水,混合均匀。
取1mL上清夜,加偏磷酸巴豆酸0.2mL/mL。
其它步骤同上。
3.保留时间的确定与样品处理相同,在 1 ml标准样品中加入0.2mL偏磷酸与巴豆酸的混合样,上样测出乙酸、丙酸、丁酸、异丁酸、戊酸和异戊酸的保留时间。
4.有机酸浓度的计算通过标准样品和内标巴豆酸各自的(或浓度)和峰面积可以计算出乙酸、丙酸及丁酸等有机酸的相对校正因子,然后根据乙酸、丙酸及丁酸的重量(或浓度)与其峰面积成正比计算出各个样品中的乙酸、丙酸及丁酸浓度。

温度决定成败——浅谈气相色谱柱温对分离效果的影响色谱柱温度,不仅影响色谱过程的热力学因素,也影响传质过程的动力学因素。
柱温变化,不仅影响柱前端压力、载气流速等,更重要的是对物质的分离、分析结果带来影响。
一、气相色谱升温方式程序升温可分为线性程序升温和非线性程序升温,前者更普遍。
线性程序升温,即随时间线性变化的升温方式,可分为一阶线性程序升温和N阶线性程序升温。
对于每阶程序升温,都包含初温、程升速率、终温以及不同温度下的保持时间四个基本参数。
气相色谱恒温分析中,对化学性质相似的同类型的化合物,保留时间和沸点呈对数关系,随着保留时间增加,峰宽迅速增加,导致先流出峰相互叠加,后流出峰又因峰展宽,使检测灵敏度下降。
因此一般通过柱温程序升温来解决这个问题。
二、程序升温阶程确定那么,程序升温时,柱温N阶程序如何确定,是否N越大越好?程序升温时,在最佳分离条件下,保留时间与沸点近似成线性关系,即随着柱温的升高,峰底宽基本不变或增加很小。
程序升温中各组分均在最佳柱温下出峰,但并不是N越多越好。
气相色谱分析中,对于组分沸点范围窄、化学性质类似的样品,如同系物,可选用一阶升温;样品组分沸点范围宽、性质差异大的,应选择N阶程序升温。
N应根据化合物的多少、需要达到的分离效果、仪器的条件等各方面来选择。
三、基本分离参数优化每阶程序升温中,设置初温、程升速率和终温这三个基本参数优化分离条件,要从分离效果和分析速度两方面考虑。
对于初温,一般比样品中沸点最低的组分沸点要低,可参考低沸点组分恒温分析时的温度。
初温的选择,主要是依据低沸点组分,但要高于固定液的凝固温度。
升温速率的选择,在了解样品组分复杂程度的基础上,既要保证较小的保留时间,又要保证较大的分离度,一般在0-10℃/min之间。
终温的选择,主要根据固定相、样品组分的热稳定性和高沸点组分的沸点确定。
同样的样品组分,流出时的柱温,在毛细管柱上的温度比填充柱低,毛细管柱上的温度一般比样品的沸点约低50℃。


实验报告实验名称:气相色谱(FID)测定水体中VFA实验日期:实验地点:姓名:班级:学号:同组实验人:一、实验目的掌握气相色谱的基本原理、组成结构及作用,了解氢火焰检测器的特点和使用方法,掌握气相色谱中利用保留时间定性的方法,以及外标定量方法,了解利用极性毛细管柱测定极性有机物的注意事项。
二、实验原理1.气相色谱工作原理:是利用试样中各组份在气相和固定液液相间的分配系数不同,当汽化后的样品被载气带入色谱柱中运行时,组份就在其中的两相间进行反复多次分配,由于固定相对各组份的吸附或溶解能力不同,因此各组份在色谱柱中的运行速度就不同,经过一定的柱长后,便彼此分离,按顺序离开色谱柱进入检测器,产生的离子流讯号经放大后,在记录器上描绘出各组份的色谱峰。
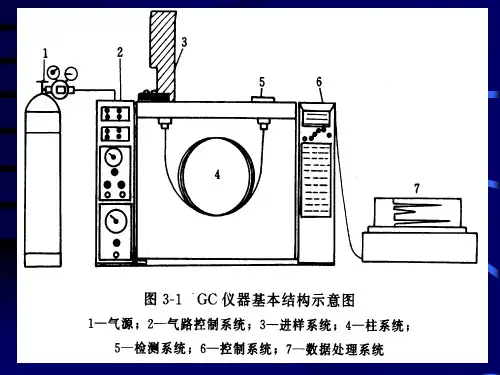
2.气相色谱仪的组成及作用:(1)载气系统:包括气源、气体净化、气体流速控制,提供稳定流量/压力的高纯载气。
(2)进样系统:包括注射器和进样口(隔垫、衬管),样品被注射器注入衬管后(液体样品将瞬间汽化),被载气带入色谱柱,分流功能也在进样口实现。
(3)色谱柱和柱温箱:在恒温或程序升温控制下,样品中各组分在色谱柱上实现分离。
(4)检测系统:获得与各组分含量呈比例的信号。
(5)记录系统:包括放大器及记录仪,或数据处理装置及工作站,记录检测器获得的信号,得到色谱图,并可以对色谱峰进行积分等处理。
3.氢火焰检测器的工作原理原理:含碳有机物在氢火焰中燃烧时,产生化学电离,发生下列反应:O H3O++CO(1)C n H m·CH;(2)·CH+O*2CHO++e-;(3)CHO++H2在电场作用下,正离子被收集到负极,产生电流。
检测器结构:如下图所示,在喷嘴上加一极化电压,氢气从管道7进入喷嘴,与载气混合后由喷嘴逸出进行燃烧,助燃空气由管道6进入,通过空气扩散器5均匀分布在火焰周围进行助燃,补充气从喷嘴管道底部8通入。
4.气相色谱保留时间定性方法在混合物样品得到分离之后,利用已知物保留值对各色谱峰进行定性是色谱法中最常用的一种定性方法。
绝对不可能,在有些场合老柱还好用,例如石油分析。
俺以前仍点文字可能对你有用。
现附上。
可从以下几个方面探讨──────────────────────────── 通用性:毛细管柱通用性好,一根非极性柱HP-1可分离诸如苯甲酸、山梨酸、BHA、BHT、TBHQ、甲醇、杂醇油、农药、车间空气等多种组分,因此可不拆柱而同时做几种实验。
填充柱通用性较差,不同项目所用柱基本不能通用。
存在着经常拆柱的麻烦。
──────────────────────────── 专用性:毛细管柱由于一般相似极性皆可出峰,因此容易产生互相干扰现象。
填充柱的特异性较强,许多干扰物质不出峰或峰形极差,干扰较小。
(例)项目:苯甲酸、山梨酸毛细管柱(HP-1)酸性及中性物质皆可出峰,出峰多填充柱(5%DEGS+1%磷酸)只有酸性物质可以出峰,出峰少──────────────────────────── 柱容量:填充柱容量较大,可以多次注入较“脏”的样品,对样品前处理要求不高。
毛细管柱容量小,且口径越小,容量越小。
如样品处理液成份复杂,极易污染柱子而影响基线及分离效果。
因此对样品前处理要求高。
──────────────────────────── 柱效能:毛细管柱明显好于填充柱,口径越细,效果越明显。
由于毛细管柱内径极小,有效地解决了柱内扩散效应,使得峰形尖锐,分离度好口径越细,效果越明显。
(例)项目:异丁醇、异戊醇毛细管柱(HP-1,HP-17)峰形尖锐填充柱(GDX-102 )峰形宽且钝。
──────────────────────────── ──────────────────────────── 灵敏度:毛细管柱稍好于填充柱。
毛细管柱的柱效高,扩散效应低,相同量的物质可以得到更高的峰高。
但由于其容量低,进样须采用分流方式,实际进入柱内的被测物往往不到10%。
──────────────────────────── 检测速度:毛细管柱优于填充柱。