病例高钾血症
- 格式:doc
- 大小:77.00 KB
- 文档页数:5

高钾血症和低钾血症危象分析与治疗(儿童常见病)高钾血症和低钾血症是儿童常见的电解质紊乱,可引起严重的心脏节律异常,危及生命。
引起血钾异常的原因很多,主要分摄入异常、细胞内外分布异常和排泄异常3类。
对高钾血症及低钾血症危象的病因、发病机制、识别与评估及干预措施进行概述。
1 钾的生理正常情况下,机体通过饮食经胃肠道摄入钾。
人体98%的钾离子在细胞内,细胞内和细胞外钾离子的浓度比约为30∶1。
该浓度梯度对维持细胞静息电位、神经肌肉兴奋性和心肌自律性十分重要。
约90%的钾离子经肾脏排出,汗液或大便排泄的钾为10%~20%。
血清钾经肾小球滤出,其中至少90%被重吸收,重吸收的主要部位是近曲小管和Henle袢。
远曲小管和集合管主动排泌钾,泌钾与摄入量、远曲小管和集合管细胞中氢离子浓度、钠离子重吸收等有关。
尿钾排泄主要通过醛固酮调节。
当机体摄入大量钾,细胞层面将对其调控,细胞膜上Na+-K+-ATP 酶(由胰岛素及α和β-2肾上腺素受体激动剂介导)使血中约80%的钾进入细胞内,以避免血清钾突然升高而危及生命,然后肾脏再泌钾,使血钾实现动态平衡,维持血清钾水平在生理浓度。
成人的血清钾浓度为3.5~5.5 mmol/L,儿童和婴儿的血清钾正常范围与年龄呈一定相关,小婴儿和早产儿的血清钾浓度上限可达6.5 mmol/L。
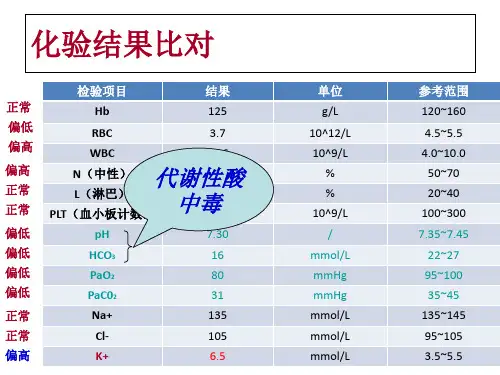
2 高钾血症危象2.1 概述儿童高钾血症定义为血清钾超过5.5 mmol/L,6~7 mmol/L为中度高钾血症,>7 mmol/L为严重高钾血症。
高钾血症危象是指伴有心电图改变(通常不包括单纯T 波改变)、肌肉无力或血钾>7 mmol/L的高钾血症。
由于血钾可持续快速升高,当细胞内钾持续释放(肿瘤溶解综合征、严重挤压伤所致横纹肌溶解)、血钾在6~7 mmol/L时,也应被视为高钾血症危象。
2.2 病因及发病机制高钾血症主要原因是钾离子从细胞内向细胞外转移,或肾排钾功能受损。
细胞内钾外移可致一过性高钾血症,而肾功能受损则导致持续高钾血症。


高钾血症的治疗措施高钾血症是指体内钾离子浓度异常升高,超过正常范围。
正常情况下,细胞内外的钾离子浓度保持一定的平衡,而高钾血症可能造成心脏等重要器官功能障碍,甚至危及生命。
因此,及时进行治疗至关重要。
本文将介绍高钾血症的治疗措施。
治疗目标高钾血症的治疗旨在降低血清钾离子浓度至正常范围内。
一般来说,治疗目标的血清钾浓度应在3.5-5.0 mmol/L范围内。
治疗措施1.危重病例处理:对于高钾血症导致心脏病变的危重病例,应迅速建立静脉通道,进行心电监护,及时进行紧急处理,如血液透析或钾离子的迅速排除等。
2.消除钾的摄入来源:避免摄入高钾食物,如蛋黄、菠菜、芋头等。
此外,停用引起高钾血症的药物,如钾盐补充剂、非甾体类抗炎药等。
3.促进钾排出:采用药物或非药物手段促进体内钾的排出。
–药物治疗:常用的药物包括利尿剂、葡萄糖和胰岛素、碳酸氢钠等。
–利尿剂:通过增加尿液排出来减少体内钾的含量。
常用的利尿剂包括袢利尿剂(如呋塞米)、噻嗪类利尿剂(如氢氯噻嗪)和钾保留性利尿剂(如螺内酯)。
具体的药物使用需根据患者具体情况和医生的建议进行选择。
–葡萄糖和胰岛素:胰岛素可调动体细胞对钾的摄取,而葡萄糖能够增加胰岛素的分泌。
因此,静脉注射葡萄糖和胰岛素可加速钾的排出。
–碳酸氢钠:碳酸氢钠可以使血液呈碱性,促进钾从细胞内转移到细胞外,并随尿液排出体外。
4.促进钾转移:碱性负荷和胰岛素可促进钾从细胞内转移到细胞外,从而减少体内钾的含量。
碳酸盐类药物可以提供碱性负荷,而胰岛素可以通过胰岛素泵或注射的方法进行补充。
5.进一步处理原发病:高钾血症常常是其他疾病的结果,例如肾功能不全、酸碱平衡失调等。
因此,在治疗高钾血症的同时,也需要针对原发病进行适当的治疗。
高钾血症的预防除了及时治疗,还应采取预防措施,包括:•确保摄入适量的钾:合理饮食,控制钾的摄入,避免过量摄入高钾食物。
•定期监测血清钾浓度:定期进行血清钾浓度检测,及时发现高钾血症,并采取相应措施。




高钾血症在临床中,高钾血症虽较低钾血症少见,但也常可发生。
高钾血症多血钾高于5.5mmol/L,即称为高钾血症,但血钾增高,并不反映全身总体钾含量增高,如单纯性脱水,有时总体钾缺乏时,血清钾也可增高,其他电解质紊乱也可影响高血钾的发生和发展。
钾是神经-肌肉兴奋性因子,而对心肌是抑制因子,因此,血钾增高,可提高神经-肌肉的兴奋,而降低心肌肉的兴奋性。
正常情况下,每日由食物中可摄入50~100 mmol/L的K+,如肾功能正常,可由尿排出进行调节,因此,不会出现血钾升高。
血钾增高可无明显临床症状,或被发病临床症状所掩盖,因此应根据疾病性质,血钾测定和心电图检查进行判断。
高钾血症是属于危急症之一,常可因血钾突然增高,但临床症状不明显,却可使心脏突然停搏而死亡,因此,应特别注意。
一、发病原因临床中可引起高钾血症的原因很多,也很复杂,常常是继发于其他疾病,特别是一些危重抢救或终末期病上,由于病情严重,合并症并发症多,而且使用药物多,常可造成肾脏损害,肾功能障碍,也是引起高钾血症的重要原因。
高钾血症发生的原因有:(一)钾摄入过多在肾脏损害、肾功障碍、尿量减少的病人,如钾摄入过多,则可引起高钾血症。
1、进食含钾丰富的食物,如蔬菜、水果、肉类或腌制的咸菜。
2、长期食用低钠高钾的食盐代用品。
3、补钾时输入钾量过多或速度过快,特别是误将10%氯化钾直接静脉注射,或配制浓度过高,发生严重的高钾血症,甚至引起心脏骤停,导致死亡,临床也有报告。
4、输入大量的含钾制剂,如静霉素钾或羧苄青霉素钾盐。
每100万单位青霉G钾盐含钾65mg(1.67mmol),相当于氯化钾125mg中钾的含量。
因钾含量较少,因此,只有大剂量(如1千万单位以上)应用或快速静脉输入时,才会使血钾明显增高,引起高钾血症。
5、服用储钾利尿剂,如氨体舒通、氨苯碟啶、吡咪嗪,可因抑制远曲小管分泌K+而引起血钾增高,或长期补充钾盐,如补充的钾超过肾脏排钾的能力,或肾小管分泌K+功能障碍,也可引起血钾增高。


最完整的高钾血症病因诊断及处理方式高钾血症对机体的主要威胁是心脏抑制,可导致严重心律失常,有些患者可无症状而突然出现心脏骤停.治疗原则是保护心脏,迅速降血钾,RF患者血钾﹥6--6.5mmol/L、ECG出现高钾表现(见后)、心率减慢、四肢无力、感觉异常、麻木等时,必须紧急处理.高钾血症三大思维方式一、对抗钾对心脏的抑制。
(一)钙剂:可对抗钾对心肌的毒性,可直接对抗高K对细胞膜复极的影响,而使阈电位复常。
常用10%葡萄酸钙20ml+5%葡萄糖注射液20ml慢静推.起作用甚快,1-3分钟见效。
但持续时间较短,仅30—60min。
注射后可用ECG监护,如10—20min未见效果,可再重复注射。
(有HF时不宜同时使用洋地黄)(二)碳酸氢钠1。
作用机制:(1)造成药物性碱血症,促使K+进入细胞内;(2)Na+对抗K+对心脏的的抑制作用;(3)可提高远端肾小管中钠含量,增加Na+—K+交换,增加尿钾排出量;(4)Na+升高血浆渗透压、扩容,起到稀释性降低血钾作用;(5)Na+有抗迷走神经作用,有利于提高HR。
2。
方法:用5%NaHCO3 20ml○v继以100-200ml快静滴。
用后一般5—10分钟起作用并持续到滴注完后2h。
滴注中应注意防止肺水肿(急性左心衰),合并HF者慎用.小部分病例由于注射后导致的碱血症快速产生,可诱发抽搐或手足搐搦症,此时可同时注射葡萄酸钙,或氯化钙对抗之,但NaHCO3不得与钙剂混合。
(三)GS+RI使血清K+转移至细胞内一般用25-50%GS,按每4-5gGS给予IuRI(普通胰岛素)持续静滴(50%GS 50—100ml+RI 16-12u或10%GS 500ml+RI 10u静脉快滴1h.注射开始后30min起作用,持续时间为4-6h。
通常应用上述剂量后血K+可下降0。
5-1。
2mmol/L。
必要时4—6h再重复一次)(四)选择性β受体激动剂可促进K+通入细胞内,如沙丁胺醇等。
高钾血症治疗措施的差别——孟庆义之吉白夕凡创作某急诊头晕患者,查体示心率很慢,血清钾7.0mmol/L,诊断为高钾血症,遂给予葡萄糖加胰岛素静脉滴注,此治疗合理吗?提起高钾血症的治疗,临床医生往往会想起葡萄糖加胰岛素、钙剂、碳酸氢钠及利尿等多种疗法,但是,如从时间维度来考量,这些治疗措施之间是存在很大差别的,即发生作用的时间明显分歧,故其应用的先后次序是有明确规定的。
即首先用离子对抗疗法,它能通过稳定细胞膜,而迅速发挥作用;其次,才是葡萄糖加胰岛素等疗法,它发挥作用的时间在半小时以后。
也许就在这半小时之内,患者发生心脏骤停,这个时间差就是抢救生命的关键所在。
1 概述正常的人体藉由饮食及水分的摄取来取得所需的营养,这些营养包含有糖类、蛋白质、淀粉、维生素及许多的矿物质元素。
钾离子即是人体重要的矿物质之一。
人体内每公斤体重含钾约2g,其中90%以上在细胞之内。
钾离子除了是细胞内液重要的元素外,在蛋白质合成及细胞内酶的作用都是重要的角色。
细胞外液中,钾离子与镁、钠、钙离子共同促进神经活动及肌肉的收缩。
对于维持心脏规律性跳动也具有重要作用。
1.1 钾在机体的含量和分布钾离子是机体最重要的阳离子之一,正凡人体内含钾总量约45 mmol/kg,其中98%在细胞内,主要分布于人的肌肉、肝、骨骼和红细胞内,红细胞内钾浓度约105 mmol/L,细胞外液中的钾含量仅占机体总钾量的2%,血浆(清)钾浓度为3.5 mmol/L~5.0 mmol/L,故临床上测血清钾时不克不及溶血,以免误诊。
在生理状态下,机体组织细胞膜电位与细胞内外钾钠离子浓度有关,且细胞外钾离子浓度远远低于细胞内钾离子浓度。
静息状态下,细胞膜对钾离子通透性最大,导致钾离子外流,使膜内电位酿成负值,(细胞膜对细胞内蛋白质无通透性,而且蛋白质带负电荷)处于极化状态。
故血清钾浓度微量变更,均会影响膜电位,即影响细胞的极化状态。
神经、肌肉、心肌细胞等的正常功能与膜电位有关,因此钾代谢紊乱、特别易出现与这些组织有关的临床症状。
疾病名:老年人高钾血症英文名:senile hyperkalemia缩写:别名:老年高钾血症;老年人血钾过多;老年人血内钾过多症;老年人高钾血疾病代码:ICD:E87.5概述:高钾血症是指血清钾>5.5mmol/L。
高血钾时体钾总量并不一定增多,但细胞内、外钾分布不正常。
除非肾脏本身病变,老年人肾脏功能并不降低;因此,高钾血症较低钾血症少见。
流行病学:老年人高钾血症相对比低钾血症少见,据老年人尸检病例统计,其发生率为8.8%。
但危害更大,常可致严重的心律失常和猝死。
病因:肾脏疾病仍是老年高钾血症的主要原因,如急性少尿性肾衰竭,慢性肾病合并饮食摄钾过多、活动性胃肠出血或存在低肾素性高醛固酮血症(特别是糖尿病肾病)。
老年高钾血症亦常由药物所引起,主要有保钾利尿剂、非类固醇抗炎药、血管紧张素转换酶抑制剂和β-肾上腺素能阻滞剂等。
此外尚可发生于代谢性酸中毒(如糖尿病酮症酸中毒等)和肾上腺功能不全者。
发病机制:高钾血症使神经肌肉细胞发生过度去极化,降低细胞膜静息电位、降低应激、减慢传导、缩短动作电位。
临床表现:直到心律失常前,高钾血症常没有症状,或者仅为不太明确的软弱和感觉异常。
心电图的早期改变是 T 波高尖、QTc 间期缩短(提示血钾水平>5.5mmol/L),随着血钾进一步升高,可出现结性和室性心律失常,并伴有 QRS 波群增宽和PR 间期延长,最后可发生心室颤动或心室停搏。
严重高钾血症亦可出现迟缓性肌肉麻痹。
并发症:可并发心律失常,室颤和心室停搏,严重高钾血症和可出现肢体软瘫。
实验室检查:血清钾水平>5.5mmol/L,总体钾常过多或正常;若正常,细胞内外钾分布不正常。
其他辅助检查:心电图检查:血清钾增高达到6mmol/L 以上时约有25%的患者可出现心电图的改变,血清钾达到8mmol/L 时,80%患者出现心电图改变。
血清钾达8~10mmol/L 时可出现严重的心律失常甚至心脏停搏。
高钾血症典型的心电图表现为 T 波高耸,QT 间期缩短,严重时 P 波消失、QRS 波增宽,进一步 S-T 段与T 波融合,T 波增宽,与QRS 波共同形成双相波浪形。
病例患者女性,49岁,因“高钾血症4年”被转诊至梅奥(Mayo)医院。
该患者肥胖,于20年前行胃囊带术治疗(术后发生持续性腹泻2~3年,但随后缓解),此外,其还患有纤维肌痛和双相情感障碍(未服用锂盐治疗)。
4.5年前,患者因单侧局限性肾细胞癌接受左侧肾及肾上腺切除术(术后未接受放化疗)。
术前,患者血压正常(120~140/70~90 mmHg),血钾处于正常高限水平(4.6~5.6 mmol/L)并被要求限制饮食钾摄入。
术后数月,患者开始自觉全身乏力,并出现轻度发作性头痛,于当地医院就诊,检查显示其处于低血压(90~95/50~56 mmHg)和高钾血症(血钾6.8 mmol/L)状态。
患者随即被送至当地急诊,接受静脉补盐和口服聚磺苯乙烯治疗。
出院时,患者服用维持剂量的盐片和聚磺苯乙烯。
其后,患者多次因类似发作入当地急诊,并接受相同治疗。
约2年前,患者出现发作性心悸伴眩晕(几乎晕厥),被诊断为阵发性室上性心动过速(PSVT)。
行经胸超声心动图检查,未发现心脏体积和功能异常;且除临界性低血压和高钾血症外,患者并无其他异常。
因此,其PSVT可能与低血压和高钾血症有关。
患者曾尝试药物治疗,但无法耐受,故转诊至梅奥医院心脏病科,接受PSVT消融治疗。
治疗前,患者接受了静脉补盐和强化聚磺苯乙烯治疗,以纠正其低血压和高钾血症;治疗后,患者被转至肾脏病科接受进一步诊治。
用药患者服用的药物包括阿普唑仑、氢可酮-对乙酰氨基酚、拉莫三嗪、氯化钠、聚磺苯乙烯和维生素A、D、E。
体格检查体质指数(BMI)30 kg/m2,呼吸18次/分,血压95/49 mmHg,脉搏89次/分,肺部听诊清晰,腹部无触痛,无坠积性水肿,无皮疹及黏膜破损。
实验室检查血细胞计数正常,血钠142 mmol/L,钾6.7 mmol/L,氯106 mmol/L,碳酸氢盐29 mmol/L,尿素氮(BUN)6.8 mmol/L(19 mg/dl),肌酐70.7 μmol/L(0.8 mg/dl),钙2.4 mmol/L(9.5 mg/dl),促甲状腺激素(TSH)2.2 mIU/L,上午皮质醇359.3 nmol/L(13 μg/dl),促皮质素 2.42 pmol/L(11 pg/ml),肾素活性 1.4 ng/(ml·h),醛固酮172 pmol/L (6.2 ng/dl)。
尿钠125 mmol/L,钾39 mmol/L,渗透压460 mOsm/kg。
提问1. 患者为何出现持续性高钾血症?2. 该患者的诊断是什么?3. 如何治疗该患者?回答下期本栏目将提供该病例的详细诊疗分析。
[3311002]49岁女性,患持续性高钾血症,4年前曾行左肾及左肾上腺切除术。
在随后3.5年中,患者常自觉全身无力并出现发作性眩晕(几乎晕厥)。
在当地医院检查示,血容量不足和高钾血症。
患者接受周期性血容量重建和标准剂量聚磺苯乙烯治疗。
转诊至梅奥医院时,患者血压为正常值低限。
实验室检查示,血钾6.7 mmol/L,肾素活性1.4 ng/(ml·h),醛固酮172 pmol/L(6.2 ng/dl);尿钠125 mmol/L,尿钾39 mmol/L,尿渗透压470 mOsm/kg。
转诊医师提问1. 患者为何出现持续性高血钾?2. 患者的诊断是什么?3. 如何治疗该患者?病情分析及追踪此患者可疑为肾素及醛固酮相对缺乏,而非醛固酮抵抗。
对患者肾小管钾浓度梯度(TTKG)进行连续检测,结果示,其基础TTKG为3.6。
予氟氢可的松0.1 mg口服,患者的TTKG迅速上升至6.2,这与醛固酮相对缺乏的诊断一致。
患者开始接受0.1 mg/d氟氢可的松补充治疗。
在随访过程中,患者血压升至108~125/65~80 mmHg(共测量6次),血钾浓度于治疗后第2周及第3个月时分别降至5.1 mmol/L和4.4 mmol/L。
最终诊断相对性低肾素性醛固酮减少症(据发生时间判断,可能与单侧肾及肾上腺切除相关)。
病因讨论钾的摄入、细胞内外转移及尿液排泄之间的平衡,决定了血钾的最终浓度。
持续性高钾血症主要发生于尿钾排泄减少,常见于以下三种情况,即肾功能衰竭、远端小管钠转运或小管液流量减少、醛固酮减少症。
肾功能衰竭通常情况下,一个成年人每日自饮食中摄入约100 mmol钾,其中90~95 mmol经肾脏排泄,5~10 mmol经胃肠道排泄。
每次饮食后,摄入的钾被快速吸收,然而血钾浓度却可维持相对稳定,其原因在于许多激素周密而快速地调节了钾的跨细胞转运。
当摄入食物后,快速升高的血胰岛素和一定程度升高的肾上腺素使新摄入的钾负荷转运至细胞内。
此后,血胰岛素和肾上腺素水平逐渐降低,细胞内钾将逐渐转移至细胞外,允许肾脏有足够的时间逐渐排泄所摄入的钾。
在6~7小时内,经饮食所摄入钾的净增加量将大部分经由肾脏排泄。
对于肾衰患者,尽管跨细胞转运可防止急性高钾血症的发生,但由于胰岛素和肾上腺素的作用在饭后数小时内逐渐减弱,因此这些患者最终仍会发生高钾血症。
尽管研究显示肾衰患者胃肠道的钾排泄能力会发生代偿性增加,但此种代偿仍远远不够。
因此,肾衰常导致慢性高钾血症。
但对于此患者而言,其肾功能尚可,因此不能以肾衰来解释其持续性高钾血症。
远端小管钠转运或小管液流量减少当肾脏有充足的肾小球滤过率(GFR)时,远端小管对钠和水的转运增加将产生电梯度和稀释性小管液,促进钾排泌。
因此,当远端小管钠转运减少和出现少尿时,肾脏对钾的排泌能力受损,导致高钾血症。
对于此患者,尽管切除了单侧肾脏,但其肾脏清除功能仍保持较好[估计的GFR为76 ml/(min·1.73m2)],因此持续性高血钾不能由其轻度的肾功能异常解释。
患者无少尿亦无尿钠浓度降低,因此这些因素也不能解释其持续性高钾血症。
醛固酮减少症低肾素性醛固酮减少症(绝对或相对)成年患者常有糖尿病、高血压和肾功能异常,血液学检查常显示为不同程度的高氯血症酸中毒、肾素活性及醛固酮浓度降低。
但回顾此患者病史,其并未出现上述已为人们熟知的改变,因此数年来其主管医师一直没有怀疑该患者是否为低肾素性醛固酮减少症。
醛固酮调节体内钠钾平衡的生理机制醛固酮在调节体内钠钾平衡中发挥了重要作用。
当肾素-血管紧张素-醛固酮系统(RAAS)受刺激或发生高钾血症时,将导致醛固酮的产生。
醛固酮可与肾单位集合管主细胞上的盐皮质激素受体(MR)结合并使之激活,进而可刺激上皮细胞钠通道(ENaC)的形成及在管腔侧的表达,最终使钠吸收增加(图1)。
钠吸收增加可导致肾小管腔内负电位产生,刺激钾和质子分泌。
体外及动物(大鼠和狗)研究显示,醛固酮也可通过上调肾小管润细胞上顶端质子泵的表达,以非钠依赖的形式直接刺激质子分泌。
醛固酮与酸碱平衡稳定尽管所有的醛固酮减少症患者都会出现某种程度的高钾血症,但并非均会发生高氯血症酸中毒(这与大鼠及狗等动物试验结果不同)。
佩雷斯(Perez)等以一组双侧肾上腺切除患者为对象的研究显示,高氯性酸中毒仅发生于肾功能存在中重度异常[GFR为15~39 ml/(min·1.73m2)]的患者,肾脏清除功能较好者[GFR为63~142 ml/(min·1.73m2)]不会发生。
论文发表于《肾单位》(Nephron 1976,17:461)杂志。
佩雷斯等进一步研究发现,尽管肾功能较好的患者不存在明显的酸中毒,但其尿铵的排泌已轻度减少,而这种减少效应在肾脏功能异常者中则被放大。
这些研究提示,醛固酮在某种程度上维持或参与了机体酸碱平衡的稳定。
当肾功能良好时,醛固酮不足对肾脏排酸的影响可获得补偿;当肾功能降低时,补偿效应被消弱,醛固酮不足所致酸排泌减少的效果将被显示出来。
此外,肾功能降低本身亦会使肾脏对碳酸氢盐的调控与再生能力减弱,导致酸中毒。
一项病例研究表明,高钾血症通过改变肾小管上皮细胞铵的产生,促使了高氯血症酸中毒的发生。
这亦显示了肾脏对酸碱平衡调节作用的整体性。
与上述临床研究一致,另外几项临床研究均显示,为醛固酮减少症患者补充外源性盐皮质激素仅能部分纠正酸血症。
总之,醛固酮如何在人体内经肾脏调节酸的排泌目前尚不完全清楚。
然而,在本病例中,我们可以认为,患者所保留的肾功能补偿了醛固酮不足对肾小管酸排泌的影响。
肾素与管球反馈和水调节此患者肾素活性较低,这与其容积不足的状态不相称。
上述状况的出现与其单侧肾切除相关,因为肾脏是循环中肾素的主要来源。
肾素是肾素-血管紧张素-醛固酮系统(RAAS)级联反应中的限速步骤。
由于低肾素血症的存在,可以预测此患者肾素下游的效应物质的水平亦应有相应下降,这其中包括效果最为强大和突出的血管紧张素Ⅱ。
众所周知,血管紧张素Ⅱ参与了管球反馈和体内水平衡的生理调节。
因此,RAAS级联/血管紧张素Ⅱ的异常将导致管球反馈和水调节的改变。
管球反馈是维持内环境稳定的负反馈环路。
当髓袢升支末端的盐浓度异常升高时,致密斑细胞将感知改变,引起入球小动脉血管收缩和肾小球滤过率(GFR)下降,从而限制远端小管失盐;当髓袢升支末端盐浓度降低时,则引起相反的效应。
管球反馈由球旁入球小动脉上的A1腺苷受体激发,并同时需要血管紧张素Ⅱ作为辅助激活因子增强管球反馈。
研究显示,血管紧张素转换酶或血管紧张素Ⅱ受体(AT1)缺陷将导致肾性失盐。
对于此例患者,其低血压和不恰当地肾性失盐可能是肾素相对缺乏所致管球反馈不足的结果。
此外,尽管存在低血压,但患者的GFR仍保持良好亦反映了管球反馈不佳致GFR下降不足。
RAAS对水平衡的调节也发挥了重要作用。
血管紧张素Ⅱ刺激口渴反应,引起水摄入增加。
在肾脏水平,RAAS可增进精氨酸加压素(A VP)介导的水潴留。
在编码肾素、血管紧张素Ⅰ转换酶、A T1(包括A T1a和A T1b)受体或单纯AT1a受体等各级RAAS系统组分基因缺陷的小鼠中,均存在多尿和不恰当肾水消耗。
至于此例患者,尽管处于低血容量状态,但在每次检查时,其血钠浓度始终维持于正常值高限(>139 mmol/L)。
这种钠浓度高于预料的可能原因为,RAAS系统缺陷致水调节能力下降。
醛固酮减少症诊断标准包括:①肾上腺糖皮质激素功能正常;②醛固酮分泌减少;③肾小管对外源性盐皮质激素反应增强。
肾小管钾浓度梯度(TTKG)对于鉴别高钾血症患者的病因是明显的醛固酮缺乏,还是相对低醛固酮血症或醛固酮抵抗很有帮助。
TTKG是皮质集合小管末端尿液与血浆间的钾浓度梯度指数(图2),其可用于估算醛固酮活性。
当高钾血症患者的TTKG<5.0时,强烈提示存在低醛固酮血症。
对于此例患者,其肾上腺糖皮质激素功能正常,但其同时存在高钾血症和基础TTKG 偏低,提示钾排泄不足。
考虑到该患者有单侧肾及肾上腺切除史,且血肾素活性相对较低,因此怀疑其为肾素及醛固酮相对不足。