卡培他滨说明书
- 格式:docx
- 大小:61.96 KB
- 文档页数:33

希罗达百科名片展开编辑本段分类及成分【分类】:乙类【治疗分类】:化学治疗【活性成分】:卡培他滨【化学名称】:5’-脱氧-5-氟-N-[(戊氧基)羰基]-胞(嘧啶核)苷【化学结构式】:【分子式】:C15H22FN3O6【分子量】:359.35【性状】: 0.15g:双凸、长方形、浅桃色包衣片,除去包衣后显白色。
一面有XELODA字样,另一面有150字样;0.5g:双凸、长方形、桃色包衣片,除去包衣后显白色。
一面有XELODA字样,另一面有500字样。
编辑本段适应症结肠癌辅助化疗:卡培他滨适用于Duke s’ C期、原发肿瘤根治术后、适于接受氟嘧啶类药物单独治疗的结肠癌患者的单药辅助治疗。
其治疗的无病生存期(DFS)不亚于5-氟尿嘧啶和甲酰四氢叶酸联合方案(5-FU/LV) 。
卡培他滨单药或与其他药物联合化疗均不能延长总生存期(OS),但已有试验数据表明在联合化疗方案中卡培他滨可较5-FU/LV改善无病生存期。
医师在开具处方使用卡培他滨单药对Dukes’ C期结肠癌进行辅助治疗时,可参考以上研究结果。
用于支持该适应症的数据来自国外临床研究(见[临床试验]部分内容)。
结直肠癌:当转移性结直肠癌患者首选单用氟嘧啶类药物治疗时,卡培他滨可用作一线化疗。
卡培他滨与其他药物联合化疗时,生存期优于5-FU/LV单药化疗。
目前尚无证据证实卡培他滨单药化疗的生存期优势。
有关卡培他滨在联合化疗中取代5-FU/LV的安全性以及生存期优势还需进一步研究。
乳腺癌联合化疗:卡培他滨可与多西紫杉醇联合用于治疗含蒽环类药物方案化疗失败的转移性乳腺癌。
乳腺癌单药化疗:卡培他滨亦可单独用于治疗对紫杉醇及含蒽环类药物化疗方案均耐药或对紫杉醇耐药和不能再使用蒽环类药物治疗(例如已经接受了累积剂量400 mg/m阿霉素或阿霉素同类物)的转移性乳腺癌患者。
耐药的定义为治疗期间疾病继续进展(有或无初始缓解),或完成含有蒽环类药物的辅助化疗后6个月内复发。

乳腺癌是全球女性最常见的恶性肿瘤之一,严重威胁女性的生命健康。
近年来,随着医学技术的不断发展,乳腺癌的治疗方法也日益丰富。
卡培他滨(Capecitabine)作为一种口服的5-氟尿嘧啶(5-FU)前药,在乳腺癌治疗中具有重要作用。
本文将详细介绍卡培他滨治疗乳腺癌的方案。
二、卡培他滨的药理作用及作用机制1. 药理作用卡培他滨是一种口服的5-氟尿嘧啶前药,经肝脏和肿瘤细胞中的酶转化为5-氟尿嘧啶,发挥抗肿瘤作用。
卡培他滨具有以下药理作用:(1)抑制DNA合成:5-氟尿嘧啶是一种胸苷酸合成酶抑制剂,可抑制肿瘤细胞DNA的合成,导致肿瘤细胞死亡。
(2)抑制RNA合成:5-氟尿嘧啶还可抑制肿瘤细胞RNA的合成,进一步影响肿瘤细胞生长。
(3)诱导肿瘤细胞凋亡:卡培他滨可诱导肿瘤细胞凋亡,发挥抗肿瘤作用。
2. 作用机制卡培他滨在肿瘤细胞中通过以下途径发挥抗肿瘤作用:(1)通过肿瘤细胞中的胸苷酸合成酶转化为5-氟尿嘧啶,抑制肿瘤细胞DNA和RNA的合成。
(2)通过增加肿瘤细胞中DNA损伤,诱导肿瘤细胞凋亡。
(3)通过抑制肿瘤细胞增殖,降低肿瘤负荷。
三、卡培他滨治疗乳腺癌的适应症及禁忌症1. 适应症(1)转移性乳腺癌:卡培他滨可用于转移性乳腺癌患者的治疗,可单独使用或与其他药物联合使用。
(2)晚期乳腺癌:对于晚期乳腺癌患者,卡培他滨可用于一线或二线治疗。
(3)辅助治疗:对于早期乳腺癌患者,卡培他滨可用于辅助治疗,降低复发风险。
(1)对卡培他滨或其活性代谢产物过敏者。
(2)严重肝、肾功能不全者。
(3)骨髓抑制:白细胞计数低于3.0×10^9/L、血小板计数低于75×10^9/L者。
(4)活动性消化道溃疡。
(5)严重感染。
四、卡培他滨治疗乳腺癌的方案1. 剂量及用法(1)转移性乳腺癌:推荐剂量为每日2.5g/m²,分两次口服,连续服用21天,休息7天。
(2)晚期乳腺癌:推荐剂量同转移性乳腺癌。

卡培他滨片2篇卡培他滨片(一)卡培他滨片是一种非处方药,主要成分为卡培他滨。
卡培他滨属于核苷类药物,具有抗病毒和抗肿瘤活性。
卡培他滨片可以用于治疗肝母细胞瘤、肺癌和结直肠癌等多种肿瘤疾病。
卡培他滨片的使用方法是口服,一般情况下每日一次。
剂量应根据患者的年龄、体重和肝功能等因素进行调整。
对于肝功能受损的患者,剂量需要进行适当减少。
卡培他滨片的治疗过程中需要注意一些事项。
首先,卡培他滨片可能会导致胃肠道反应,如恶心、呕吐和腹泻等。
在使用期间,患者应注意饮食调理,避免食用刺激性食物,保持良好的饮食习惯。
其次,卡培他滨片还可能会抑制骨髓造血功能,导致白细胞、血小板和红细胞等下降。
因此,患者应及时检查血常规,确保白细胞、血小板和红细胞等指标在正常范围内。
此外,还需要注意肝功能和肾功能的监测,及时发现并处理异常情况。
卡培他滨片作为药物,虽然具有明显的抗病毒和抗肿瘤活性,但是在使用过程中也存在不可忽视的不良反应。
比如,卡培他滨片可能会导致皮疹、过敏反应和肝损伤等。
因此,在使用卡培他滨片期间,患者应密切关注身体状况,一旦出现不适应立即停药,并告知医生进行处理。
总之,卡培他滨片作为一种非处方药,具有一定的疗效,但在使用过程中需要谨慎,遵循医生的指导和药品使用说明,以减少不良反应的发生,并达到更好的治疗效果。
卡培他滨片(二)卡培他滨片是一种抗肿瘤药物,其主要成分为卡培他滨。
卡培他滨通过抑制DNA合成,从而阻断了癌细胞的生长和扩散,可以用于治疗肝母细胞瘤、肺癌和结直肠癌等多种恶性肿瘤。
卡培他滨片的使用方法非常简单,只需口服即可。
通常情况下,每日一次,剂量根据患者的具体情况而定。
在用药期间,患者需要定期进行复查,通过检查癌细胞浸润和肿瘤大小的变化来评估治疗效果。
卡培他滨片在治疗肿瘤过程中,也会产生一些副作用。
常见的副作用包括恶心、呕吐、食欲减退和腹泻等胃肠道反应。
为了减轻这些不良反应,患者可以适当调整饮食,避免辛辣刺激性食物的摄入,保持良好的饮食习惯。

奥沙利铂卡培他滨方案奥沙利铂卡培他滨方案简介奥沙利铂卡培他滨方案是一种用于治疗癌症的综合治疗方案。
该方案主要使用了奥沙利铂和卡培他滨这两种药物,通过联合使用,达到更好的治疗效果。
奥沙利铂是一种烷化剂,能够抑制癌细胞的DNA合成,进而抑制癌细胞的生长和分裂。
卡培他滨是一种抑制细胞色素P450的药物,可以增加奥沙利铂在体内的浓度,提高治疗效果。
本文将详细介绍奥沙利铂卡培他滨方案的用药方法、副作用以及注意事项。
用药方法药物剂量- 奥沙利铂:一般剂量为每平方米体表面积125 mg,溶于5%葡萄糖中,静脉滴注2小时。
- 卡培他滨:一般剂量为每天1250mg/m²,分两次口服,连续14天。
用药周期奥沙利铂卡培他滨方案的治疗周期一般为21天,包括:- 第1天:奥沙利铂静脉滴注- 第1-14天:卡培他滨口服- 第15-21天:休息期按照这个周期,连续进行多个周期的治疗。
副作用奥沙利铂的副作用- 消化系统反应:包括恶心、呕吐、腹泻等,可通过合理的支持治疗缓解。
- 血液系统反应:包括白细胞减少、血小板减少等,需定期检查血常规,并根据具体情况调整治疗方案。
- 神经系统反应:主要表现为周围神经病变和感觉异常,可通过适当的药物治疗缓解。
卡培他滨的副作用- 消化系统反应:包括恶心、呕吐、纳差等,可通过合理的支持治疗缓解。
- 血液系统反应:包括白细胞减少、血小板减少等,需定期检查血常规。
- 肝功能损害:可能引起肝功能异常,需定期检查肝功能。
注意事项1. 在接受奥沙利铂卡培他滨方案治疗期间,要定期进行血常规、肝功能等检查,以监测药物的疗效和副作用。
2. 在用药期间,应避免接触病毒性感染,注意个人卫生。
3. 在用药期间,应避免使用有肾毒性的药物,以免加重药物的肾脏副作用。
4. 在用药期间,如出现明显的不良反应或副作用,应立即咨询医生。
5. 在用药期间,应注意饮食调理,合理搭配膳食,以增强机体的免疫力。
结论奥沙利铂卡培他滨方案是一种用于治疗癌症的综合治疗方案,通过联合使用两种药物,达到更好的治疗效果。

卡培他滨单药化疗方案卡培他滨单药化疗方案:攻克癌症的里程碑癌症是当今社会面临的重大挑战之一,而化疗则是目前最常用的治疗手段之一。
卡培他滨作为一种新型的单药化疗方案,近年来备受瞩目。
本文将重点介绍卡培他滨的作用机制、应用领域和药物特点,以及它在临床实践中的效果。
卡培他滨,是一种核苷类似物,通过干扰DNA合成过程,抑制癌细胞的增殖和分裂。
相较于其他化疗药物,卡培他滨具有很强的选择性,可以选择性地杀死癌细胞而不损伤正常细胞。
这一点非常重要,因为常规化疗可能会对健康细胞造成一定的伤害,导致不良反应,如恶心、呕吐、脱发等。
而卡培他滨的独特作用机制可以最大限度地减少这些不良反应,提高患者的生活质量。
卡培他滨的应用领域非常广泛,可以用于治疗多种癌症,如结直肠癌、胃癌、乳腺癌、肺癌等。
根据患者的具体情况和癌症的类型,医生可以根据需要调整卡培他滨的剂量和使用时间。
这种个体化的治疗方案可以最大限度地发挥药物的疗效,提高患者的生存率。
除了广泛的应用领域,卡培他滨还具有一些独特的药物特点,值得我们关注。
首先,卡培他滨口服方便,避免了注射带来的痛苦和不适。
其次,卡培他滨具有较短的半衰期,即它在体内的消失速度较快,有效地减少了药物在体内的堆积风险。
这也允许医生更灵活地调整药物的剂量和方案。
此外,卡培他滨也被证明具有良好的耐受性,即使在长时间的应用下,也能减少不良反应的风险。
在临床实践中,卡培他滨的效果显著,为癌症患者带来了新的希望。
研究表明,卡培他滨不仅可以延长患者的生存时间,还能显著提高患者的生存率。
一项针对晚期结直肠癌的研究发现,卡培他滨联合其他化疗药物的治疗方案,与传统的化疗方案相比,能够显著提高患者的生存率,并延长无进展生存时间。
这一发现引起了广泛的关注,并获得了学术界的高度评价。
然而,我们也应该看到卡培他滨作为一种新型药物,仍然存在一些潜在的问题。
首先,卡培他滨的价格较高,对于一些经济困难的患者来说,可能是一个不可承受的负担。

卡培他滨 (Capecitabine) 抗癌药物卡培他滨(Capecitabine)抗癌药物卡培他滨(Capecitabine)是一种重要的抗癌药物,广泛应用于临床治疗多种恶性肿瘤。
它属于一类叫做口服脱氧尿苷类似物(Oral Fluoropyrimidines)的化学药物,它通过被转化为5-氟尿嘧啶(5-Fluorouracil,5-FU),使细胞内的嘧啶核苷酸合成减少,从而抑制了DNA的合成,达到抑制癌细胞生长和繁殖的效果。
本文将详细介绍卡培他滨的药物特性、临床应用、副作用以及药物相互作用等方面内容。
一、药物特性卡培他滨是一种咪唑型衍生物,在体内通过肝脏中的酶系反应转化为活性代谢产物,进而发挥药效。
其特点在于其制剂为口服制剂,使得患者可以在家中自行服用,大大提高了患者的治疗便利性和生活质量。
二、临床应用卡培他滨主要用于治疗结直肠癌、乳腺癌、胃癌以及胰腺癌等多种恶性肿瘤。
在结直肠癌的治疗中,卡培他滨可以单药应用或与其他化疗药物联合应用。
在乳腺癌的治疗中,卡培他滨通常与他莫昔芬(Tamoxifen)等内分泌治疗药物联合应用。
临床试验证明,卡培他滨的应用可以有效抑制肿瘤的生长和转移,并提高患者的生存率。
三、副作用卡培他滨的主要副作用包括消化系统反应、血液系统反应、皮肤反应、神经系统反应和肝功能受损等。
消化系统反应主要表现为恶心、呕吐和腹泻等症状;血液系统反应主要表现为骨髓抑制,导致贫血、白细胞减少和血小板减少等;皮肤反应主要为手足综合征,表现为手掌脱皮、疼痛和红肿等;神经系统反应主要为感觉异常和周围神经病变等。
此外,卡培他滨的应用还可能导致肝功能受损。
四、药物相互作用卡培他滨与其他药物之间存在一定的相互作用。
一些药物可能增强或降低卡培他滨的药效,例如抗血小板药物、抗凝药物和抗癫痫药物等。
因此,在患者接受卡培他滨治疗时,需告知医生目前正在使用的其他药物,以避免不良的药物相互作用。
总结:卡培他滨(Capecitabine)是一种口服的抗癌药物,通过转化为活性代谢产物抑制嘧啶核苷酸的合成,从而抑制癌细胞的生长和繁殖。

卡培他滨(Capecitabine):一种口服化疗药物的详细介绍
摘要:本文将详细介绍卡培他滨(Capecitabine),这是一种口服化疗药物。
卡培他滨可以通过肝脏代谢转化为5-氟尿嘧啶,在体内释放出活性成分,使用便捷且广泛应用于多种癌症的治疗,如结直肠癌、乳腺癌和胃癌等。
我们将探讨卡培他滨的作用机制、药理特点、临床应用、副作用以及注意事项。
1. 作用机制:
-卡培他滨通过与癌细胞内的酶(胸腺嘧啶钷酶)相互作用,将其转化为5-氟尿嘧啶,进而抑制DNA合成和细胞增殖。
-它主要通过阻断癌细胞的新生血管形成、激活免疫细胞和诱导细胞凋亡等机制来抗癌。
2. 药理特点:
-卡培他滨是一种口服药物,被肠道吸收后通过肝脏代谢转化为5-氟尿嘧啶。
-它在体内能够广泛分布到全身组织器官,并在肿瘤部位释放出活性成分。
3. 临床应用:
-卡培他滨被广泛用于多种癌症类型的治疗,包括但不限于:
-结直肠癌的早期和晚期治疗;
-乳腺癌的早期和晚期治疗;
-胃癌和食管癌等其他消化系统癌症的治疗。
4. 副作用:
-卡培他滨的常见副作用包括恶心、呕吐、腹泻、口腔溃疡、疲劳等。
-稀有但严重的副作用可能包括心脏毒性、肝功能异常和神经毒性等。
5. 注意事项:
-使用卡培他滨需在严密的医疗监督下进行,并需遵循相关的用药指导和剂量调整。
-患者在治疗期间需要定期进行血常规、肝功能监测和口腔检查。
-在使用卡培他滨期间,患者需告知医生关于任何新出现的不适或副作用。

卡培他滨用药教育
1.用法用量:
卡培他滨用药以3周为一个疗程,治疗2周后停药1周。
卡培他滨片剂应在餐后30分钟内用水吞服,每日2次。
该药物用量需根据您的体重身高综合出的体表面积计算得出,因此请在每次入院时准确称量您的身高体重并向护理人员告知。
【药学提示】
⑴为预防卡培他滨引起的手足综合征,用药同时口服vitB6 每天3次,每次30mg 。
2.常见不良反应
用药后可能会出现恶心、呕吐、腹泻、骨髓抑制、手足综合征、口腔炎等,如出现不适症状请随时与管床医师或药师联系。
【药学提示】
⑴用药期间患者应避免手足频繁摩擦和过度挤压,避免较重的体力劳动及激烈的运动。
3.出院用药指导:
⑴需要使用重组人粒细胞集落刺激因子时与化疗药物至少间隔24小时。
⑵院外请注意复查血常规,每周至少检查2次。
如白细胞<3.0×109/L或中性粒细胞<1.5×109/L时,应停服卡培他滨,予以升白治疗;待白细胞恢复后再行服用,期间漏服卡培他滨不补服。
如有异常请及时复诊。

卡培他滨单药化疗方案引言卡培他滨(Capecitabine)是一种口服的细胞毒性药物,广泛用于多种癌症的治疗,包括结直肠癌、乳腺癌和胃癌等。
本文将详细介绍卡培他滨单药化疗方案的使用方法、副作用以及注意事项。
卡培他滨的作用卡培他滨是一种鸟苷类似物,通过被癌细胞内的三磷酸化酶转化为活性代谢产物,抑制DNA合成和细胞增殖,从而达到治疗癌症的效果。
使用方法卡培他滨常用的剂量是1250mg/m2 每日口服两次,连续14天,然后休息7天。
这个14天的治疗周期被称为一个疗程。
根据癌症的具体类型和患者的情况,医生可能会调整剂量和疗程的长度。
卡培他滨应该在饭后30分钟内口服,并且应该用足够的水将药片吞下去。
如果患者无法吞咽整个药片,可以将药片细碎、搅拌到非碳酸饮料中,并立即饮用。
在服药期间,患者应该避免吃含有柠檬酸的食物,因为柠檬酸会影响卡培他滨的吸收。
副作用卡培他滨单药化疗方案可能会引起一系列副作用,这些副作用的严重程度和持续时间因个体而异。
以下是一些常见的副作用:1.恶心和呕吐:患者常常会在治疗开始后几小时内出现恶心和呕吐的症状。
在医生的指导下,可以使用抗恶心药物来减轻这些症状。
2.腹泻:卡培他滨可能导致腹泻,有些患者可能需要使用止泻药来缓解症状。
3.手足综合征:手足综合征是卡培他滨比较特殊的副作用,表现为手掌和脚底皮肤红肿、脱皮、疼痛等症状。
保持双手和双脚的卫生,并使用滋润剂来缓解症状。
4.疲劳和乏力:卡培他滨可能导致患者感到疲劳和乏力,这时可以适当休息,提高饮食及补充营养,合理安排活动。
5.血液问题:卡培他滨可能会导致血小板和白细胞减少,使患者更容易出血和感染。
如果出现严重的血液问题,医生可能会调整剂量或延长疗程之间的休息时间。
注意事项在使用卡培他滨单药化疗方案时,患者需要注意以下几点事项:1.遵循医生的指导:卡培他滨是一种强效药物,必须按照医生的指示使用,不得随意改变剂量或停药。
2.定期复查:患者在接受卡培他滨治疗期间,需要定期进行血液检查和其他相关检查,以监测药物的疗效和副作用。
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use XELODA® safely and effectively. See full prescribing information for XELODA®.XELODA® (capecitabine) Tablets, Film Coated for Oral useInitial U.S. Approval: 1998WARNING: XELODA-WARFARIN INTERACTIONSee full prescribing information for complete boxed warning. Patients receiving concomitant XELODA and oral coumarin-derivative anticoagulants such as warfarin and phenprocoumon should have their anticoagulant response (INR or prothrombin time) monitored frequently in order to adjust the anticoagulant dose accordingly. Altered coagulation parameters and/or bleeding, including death, have been reported during concomitant use.• Occurrence: Within several days and up to several months after initiating XELODA therapy; may also be seen within 1 month after stopping XELODA• Predisposing factors: age>60 and diagnosis of cancer---------------------------INDICATIONS AND USAGE----------------------------XELODA (capecitabine) is a nucleoside metabolic inhibitor with antineoplastic activity indicated for:• Adjuvant Colon Cancer (1.1)– Patients with Dukes’ C colon cancer• Metastatic Colorectal Cancer (1.1)– First-line as monotherapy when treatment with fluoropyrimidine therapy alone is preferred• Metastatic Breast Cancer (1.2)– In combination with docetaxel after failure of prior anthracyclinecontaining therapy– As monotherapy in patients resistant to both paclitaxel and an anthracycline-containing regimen-----------------------DOSAGE AND ADMINISTRATION -----------------------• Take XELODA with water within 30 min after a meal (2)• Monotherapy: 1250 mg/m2 twice daily orally for 2 weeks followed by a one week rest period in 3-week cycles (2.1)• Adjuvant treatment is recommended for a total of 6 months (8 cycles)(2.1)• In combination with docetaxel, the recommended dose of XELODA is 1250 mg/m2 twice daily for 2 weeks followed by a 7-day rest period,combined with docetaxel at 75 mg/m2 as a 1-hour IV infusion every 3weeks (2.1)• XELODA dosage may need to be individualized to optimize patient management (2.2)• Reduce the dose of XELODA by 25% in patients with moderate renal impairment (2.3)---------------------DOSAGE FORMS AND STRENGTHS----------------------• Tablets: 150 mg and 500 mg (3) ------------------------------CONTRAINDICATIONS ------------------------------•Dihydropyrimidine dehydrogenase (DPD) deficiency (4.1)•Severe Renal Impairment (4.2)•Hypersensitivity (4.3)-----------------------WARNINGS AND PRECAUTIONS -----------------------• Diarrhea: May be severe. Interrupt XELODA treatment immediately until diarrhea resolves or decreases to grade 1. Recommend standardantidiarrheal treatments. (5.1)• Coagulopathy: May result in bleeding, death. Monitor anticoagulant response (e.g., INR) and adjust anticoagulant dose accordingly. (5.2) • Cardiotoxicity: Common in patients with a prior history of coronary artery disease. (5.3)• Pregnancy: Can cause fetal harm. Advise women of the potential risk to the fetus. (5.6, 8.1)• Hand-and-Foot Syndrome (Grade 2 or 3): Interrupt XELODA treatment until the event resolves or decreases in intensity. (5.7)• Hyperbilirubinemia (Grade 2 to 4): Interrupt XELODA treatment immediately until the hyperbilirubinemia resolves or decreases inintensity. (5.8)• Hematologic: Do not treat patients with neutrophil counts <1.5 x 109/L or thrombocyte counts <100 x 109/L. If grade 3-4 neutropenia orthrombocytopenia occurs, stop therapy until condition resolves. (5.9)------------------------------ADVERSE REACTIONS ------------------------------Most common adverse reactions (≥30%) were diarrhea, hand-and-foot syndrome, nausea, vomiting, abdominal pain, fatigue/weakness, and hyperbilirubinemia. Other adverse reactions, including serious adverse reactions, have been reported. (6)To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or /medwatch------------------------------DRUG INTERACTIONS-------------------------------• Anticoagulants: Monitor anticoagulant response (INR or prothrombin time) frequently in order to adjust the anticoagulant dose as needed. (5.2,7.1)• Phenytoin: Monitor phenytoin levels in patients taking XELODA concomitantly with phenytoin. The phenytoin dose may need to bereduced. (7.1)• Leucovorin: The concentration of 5-fluorouracil is increased and its toxicity may be enhanced by leucovorin. (7.1)• CYP2C9 substrates: Care should be exercised when XELODA is coadministered with CYP2C9 substrates. (7.1)• Food reduced both the rate and extent of absorption of capecitabine. (2,7.1, 12.3)-----------------------USE IN SPECIFIC POPULATIONS -----------------------• Nursing Mothers: Discontinue nursing when receiving XELODA treatment. (8.3)• Geriatric: Greater incidence of adverse reactions. Monitoring required.(8.5)• Hepatic Impairment: Monitoring is recommended in patients with mild to moderate hepatic impairment. (8.6)• Renal Impairment: Reduce XELODA starting dose in patients with moderate renal impairment (2.3, 8.7, 12.3)See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labelingRevised: [m/year]______________________________________________________________________________________________________________________________________FULL PRESCRIBING INFORMATION: CONTENTS*WARNING: XELODA-WARFARIN INTERACTION1 .. INDICATIONS AND USAGE1.1 Colorectal Cancer1.2 Breast Cancer2 .. DOSAGE AND ADMINISTRATION2.1 Standard Starting Dose2.2 Dose Management Guidelines2.3 Adjustment of Starting Dose in Special Populations3 .. DOSAGE FORMS AND STRENGTHS4 .. CONTRAINDICATIONS4.1 Dihydropyrimidine Dehydrogenase (DPD) Deficiency4.2 Severe Renal Impairment4.3 Hypersensitivity5 .. WARNINGS AND PRECAUTIONS5.1 Diarrhea5.2 Coagulopathy5.3 Cardiotoxicity5.4 Dihydropyrimidine Dehydrogenase Deficiency5.5 Renal Insufficiency5.6 Pregnancy5.7 Hand-and-Foot Syndrome5.8 Hyperbilirubinemia5.9 Hematologic5.10 Geriatric Patients5.11 Hepatic Insufficiency5.12 Combination With Other Drugs6 .. ADVERSE REACTIONS6.1 Adjuvant Colon Cancer6.2 Metastatic Colorectal Cancer6.3 Breast Cancer6.4 Clinically Relevant Adverse Events in <5% of Patients7 .. DRUG INTERACTIONS7.1 Drug-Drug Interactions7.2 Drug-Food Interaction8 .. USE IN SPECIFIC POPULATIONS8.1 Pregnancy: Category D8.3 Nursing Mothers8.4 Pediatric Use8.5 Geriatric Use8.6 Hepatic Insufficiency8.7 Renal Insufficiency10 . OVERDOSAGE11 . DESCRIPTION12 . CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.3 Pharmacokinetics13 . NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility14 . CLINICAL STUDIES14.1 Adjuvant Colon Cancer14.2 Metastatic Colorectal Cancer14.3 Breast Cancer15 . REFERENCES16 . HOW SUPPLIED/STORAGE AND HANDLING17 . PATIENT COUNSELING INFORMATION17.1 Information for Patients (see Patient Package Insert)17.2 Patient Package Insert*Sections or subsections omitted from the full prescribing information are not listed______________________________________________________________________________________________________________________________________ FULL PRESCRIBING INFORMATIONWARNING: XELODA-WARFARIN INTERACTIONXELODA Warfarin Interaction: Patients receiving concomitant capecitabine and oral coumarin-derivative anticoagulant therapy should have their anticoagulant response (INR or prothrombin time) monitored frequently in order to adjust the anticoagulant dose accordingly. A clinically important XELODA-Warfarin drug interaction was demonstrated in a clinical pharmacology trial [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. Altered coagulation parameters and/or bleeding, including death, have been reported in patients taking XELODA concomitantly with coumarin-derivative anticoagulants such as warfarin and phenprocoumon. Postmarketing reports have shown clinically significant increases in prothrombin time (PT) and INR in patients who were stabilized on anticoagulants at the time XELODA was introduced. These events occurred within several days and up to several months after initiating XELODA therapy and, in a few cases, within 1 month after stopping XELODA. These events occurred in patients with and without liver metastases. Age greater than 60 and a diagnosis of cancer independently predispose patients to an increased risk of coagulopathy.ANDUSAGE1 INDICATIONS1.1 Colorectal Cancer• XELODA is indicated as a single agent for adjuvant treatment in patients with Dukes’ C colon cancer who have undergone complete resection of the primary tumor when treatment with fluoropyrimidine therapy alone is preferred. XELODA was non-inferior to 5-fluorouracil and leucovorin (5-FU/LV) for disease-free survival (DFS). Physicians should consider results of combination chemotherapy trials, which have shown improvement in DFS and OS, when prescribing single-agent XELODA in the adjuvant treatment of Dukes’C colon cancer.• XELODA is indicated as first-line treatment of patients with metastatic colorectal carcinoma when treatment with fluoropyrimidine therapy alone is preferred. Combination chemotherapy has shown asurvival benefit compared to 5-FU/LV alone. A survival benefit over 5-FU/LV has not been demonstrated with XELODA monotherapy. Use of XELODA instead of 5-FU/LV in combinations has not beenadequately studied to assure safety or preservation of the survival advantage.Cancer1.2 Breast• XELODA in combination with docetaxel is indicated for the treatment of patients with metastatic breast cancer after failure of prior anthracycline-containing chemotherapy.• XELODA monotherapy is also indicated for the treatment of patients with metastatic breast cancer resistant to both paclitaxel and an anthracycline-containing chemotherapy regimen or resistant to paclitaxel and for whom further anthracycline therapy is not indicated (e.g., patients who have received cumulative doses of 400 mg/m2 of doxorubicin or doxorubicin equivalents). Resistance is defined as progressive disease while on treatment, with or without an initial response, or relapse within 6 months of completing treatment with an anthracycline-containing adjuvant regimen.2 DOSAGE AND ADMINISTRATIONXELODA tablets should be swallowed whole with water within 30 minutes after a meal. XELODA dose is calculated according to body surface area.2.1 Standard Starting DoseMonotherapy (Metastatic Colorectal Cancer, Adjuvant Colorectal Cancer, Metastatic Breast Cancer)The recommended dose of XELODA is 1250 mg/m2 administered orally twice daily (morning and evening; equivalent to 2500 mg/m2 total daily dose) for 2 weeks followed by a 1-week rest period given as 3-week cycles (see Table 1).Adjuvant treatment in patients with Dukes’ C colon cancer is recommended for a total of 6 months [ie, XELODA 1250 mg/m2 orally twice daily for 2 weeks followed by a 1-week rest period, given as 3-week cycles for a total of 8 cycles (24 weeks)].Table 1 XELODA Dose Calculation According to Body Surface AreaDose Level 1250 mg/m2 Twice a Day Number of Tablets to be Taken at Each Dose (Morning and Evening)Surface Area(m2) Total DailyDose* (mg)150 mg 500 mg≤ 1.25 3000 0 31.26-1.37 3300 1 31.38-1.51 3600 2 31.52-1.65 4000 0 41.66-1.77 4300 1 41.78-1.91 4600 2 41.92-2.05 5000 0 52.06-2.17 5300 1 5≥ 2.18 5600 2 5*Total Daily Dose divided by 2 to allow equal morning and evening dosesIn Combination With Docetaxel (Metastatic Breast Cancer)In combination with docetaxel, the recommended dose of XELODA is 1250 mg/m2 twice daily for 2 weeks followed by a 1-week rest period, combined with docetaxel at 75 mg/m2 as a 1-hour intravenous infusion every 3 weeks. Pre-medication, according to the docetaxel labeling, should be started prior to docetaxel administration for patients receiving the XELODA plus docetaxel combination. Table 1 displays the total daily dose of XELODA by body surface area and the number of tablets to be taken at each dose.2.2 Dose Management GuidelinesGeneralXELODA dosage may need to be individualized to optimize patient management. Patients should be carefully monitored for toxicity and doses of XELODA should be modified as necessary to accommodate individual patient tolerance to treatment [see Clinical Studies (14)]. Toxicity due to XELODA administration may be managed by symptomatic treatment, dose interruptions and adjustment of XELODA dose. Once the dose has been reduced, it should not be increased at a later time. Doses of XELODA omitted for toxicity are not replaced or restored; instead the patient should resume the planned treatment cycles.The dose of phenytoin and the dose of coumarin-derivative anticoagulants may need to be reduced when either drug is administered concomitantly with XELODA [see Drug Interactions (7.1)].Monotherapy (Metastatic Colorectal Cancer, Adjuvant Colorectal Cancer, Metastatic Breast Cancer) XELODA dose modification scheme as described below (see Table 2) is recommended for the management of adverse reactions.Table 2 Recommended Dose Modifications of XELODAToxicityNCIC Grades* During a Course of TherapyDose Adjustment for Next Treatment (% of starting dose)Grade 1 Maintain dose level Maintain dose level Grade 2-1st appearanceInterrupt until resolved to grade 0-1 100%-2nd appearance 75% -3rd appearance 50% -4th appearance Discontinue treatment permanently -Grade 3-1st appearanceInterrupt until resolved to grade 0-1 75%-2nd appearance 50%-3rd appearance Discontinue treatment permanently -Grade 4-1st appearance Discontinue permanentlyORIf physician deems it to be in thepatient’s best interest to continue,interrupt until resolved to grade 0-150%*National Cancer Institute of Canada Common Toxicity Criteria were used except for the hand-and-foot syndrome [see Warnings and Precautions (5)].In Combination With Docetaxel (Metastatic Breast Cancer)Dose modifications of XELODA for toxicity should be made according to Table 2 above for XELODA. At the beginning of a treatment cycle, if a treatment delay is indicated for either XELODA or docetaxel, then administration of both agents should be delayed until the requirements for restarting both drugs are met.The dose reduction schedule for docetaxel when used in combination with XELODA for the treatment of metastatic breast cancer is shown in Table 3.Table 3 Docetaxel Dose Reduction Schedule in Combination with XELODA ToxicityNCIC Grades*Grade 2 Grade 3 Grade 41st appearance Delay treatment untilresolved to grade 0-1;Resume treatment withoriginal dose of 75 mg/m2docetaxel Delay treatment untilresolved to grade 0-1;Resume treatment at55 mg/m2 of docetaxel.Discontinue treatmentwith docetaxel2nd appearance Delay treatment untilresolved to grade 0-1;Resume treatment at55 mg/m2 of docetaxel. Discontinue treatmentwith docetaxel-3rd appearance Discontinue treatment withdocetaxel--*National Cancer Institute of Canada Common Toxicity Criteria were used except for hand-and-foot syndrome [see Warnings and Precautions (5)].2.3 Adjustment of Starting Dose in Special PopulationsRenal ImpairmentNo adjustment to the starting dose of XELODA is recommended in patients with mild renal impairment (creatinine clearance = 51 to 80 mL/min [Cockroft and Gault, as shown below]). In patients with moderate renalstarting dose when used as monotherapy or in combination with docetaxel (from 1250 mg/m2 to 950 mg/m2 twice daily) is recommended [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)]. Subsequent dose adjustment is recommended as outlined in Table 2 and Table 3 (depending on the regimen) if a patient develops a grade 2 to 4 adverse event [see Warnings and Precautions (5.5)]. The starting dose adjustment recommendations for patients with moderate renal impairment apply to both XELODA monotherapy and XELODA in combination use with docetaxel.Cockroft and Gault Equation:(140 - age [yrs]) (body wt [kg])Creatinine clearance for males = —————————————(72) (serum creatinine [mg/dL])Creatinine clearance for females = 0.85 x male valueGeriatricsPhysicians should exercise caution in monitoring the effects of XELODA in the elderly. Insufficient data are available to provide a dosage recommendation.3 DOSAGE FORMS AND STRENGTHSXELODA is supplied as biconvex, oblong film-coated tablets for oral administration. Each light peach-colored tablet contains 150 mg of capecitabine and each peach-colored tablet contains 500 mg of capecitabine.4 CONTRAINDICATIONS4.1 Dihydropyrimidine Dehydrogenase (DPD) DeficiencyXELODA is contraindicated in patients with known dihydropyrimidine dehydrogenase (DPD) deficiency.4.2 Severe Renal ImpairmentXELODA is contraindicated in patients with severe renal impairment (creatinine clearance below 30 mL/min [Cockroft and Gault]) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].4.3 HypersensitivityXELODA is contraindicated in patients with known hypersensitivity to capecitabine or to any of its components. XELODA is contraindicated in patients who have a known hypersensitivity to 5-fluorouracil.5 WARNINGS AND PRECAUTIONSGeneralPatients receiving therapy with XELODA should be monitored by a physician experienced in the use of cancer chemotherapeutic agents. Most adverse reactions are reversible and do not need to result in discontinuation, although doses may need to be withheld or reduced [see Dosage and Administration (2.2)].5.1 DiarrheaXELODA can induce diarrhea, sometimes severe. Patients with severe diarrhea should be carefully monitored and given fluid and electrolyte replacement if they become dehydrated. In 875 patients with either metastatic breast or colorectal cancer who received XELODA monotherapy, the median time to first occurrence of grade 2 to 4 diarrhea was 34 days (range from 1 to 369 days). The median duration of grade 3 to 4 diarrhea was 5 days. National Cancer Institute of Canada (NCIC) grade 2 diarrhea is defined as an increase of 4 to 6 stools/day or nocturnal stools, grade 3 diarrhea as an increase of 7 to 9 stools/day or incontinence and malabsorption, and grade 4 diarrhea as an increase of ≥10 stools/day or grossly bloody diarrhea or the need for parenteral support. If grade 2, 3 or 4 diarrhea occurs, administration of XELODA should be immediately interrupted until theoccurrence of any grade 3 or 4 diarrhea, subsequent doses of XELODA should be decreased [see Dosage and Administration (2.2)]. Standard antidiarrheal treatments (eg, loperamide) are recommended.Necrotizing enterocolitis (typhlitis) has been reported.5.2 CoagulopathyPatients receiving concomitant capecitabine and oral coumarin-derivative anticoagulant therapy should have their anticoagulant response (INR or prothrombin time) monitored closely with great frequency and the anticoagulant dose should be adjusted accordingly [see Boxed Warning and Drug Interactions (7.1)].5.3 CardiotoxicityThe cardiotoxicity observed with XELODA includes myocardial infarction/ischemia, angina, dysrhythmias, cardiac arrest, cardiac failure, sudden death, electrocardiographic changes, and cardiomyopathy. These adverse reactions may be more common in patients with a prior history of coronary artery disease.5.4 Dihydropyrimidine Dehydrogenase DeficiencyRarely, unexpected, severe toxicity (eg, stomatitis, diarrhea, neutropenia and neurotoxicity) associated with 5fluorouracil has been attributed to a deficiency of dihydropyrimidine dehydrogenase (DPD) activity. A link between decreased levels of DPD and increased, potentially fatal toxic effects of 5-fluorouracil therefore cannot be excluded.5.5 Renal InsufficiencyPatients with moderate renal impairment at baseline require dose reduction [see Dosage and Administration (2.3)]. Patients with mild and moderate renal impairment at baseline should be carefully monitored for adverse reactions. Prompt interruption of therapy with subsequent dose adjustments is recommended if a patient develops a grade 2 to 4 adverse event as outlined in Table 2 [see Dosage and Administration (2.2), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].5.6 PregnancyXELODA may cause fetal harm when given to a pregnant woman. Capecitabine caused embryolethality and teratogenicity in mice and embryolethality in monkeys when administered during organogenesis. If this drug is used during pregnancy, or if a patient becomes pregnant while receiving XELODA, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].5.7 Hand-and-Foot SyndromeHand-and-foot syndrome (palmar-plantar erythrodysesthesia or chemotherapy-induced acral erythema) is a cutaneous toxicity. Median time to onset was 79 days (range from 11 to 360 days) with a severity range of grades 1 to 3 for patients receiving XELODA monotherapy in the metastatic setting. Grade 1 is characterized by any of the following: numbness, dysesthesia/paresthesia, tingling, painless swelling or erythema of the hands and/or feet and/or discomfort which does not disrupt normal activities. Grade 2 hand-and-foot syndrome is defined as painful erythema and swelling of the hands and/or feet and/or discomfort affecting the patient’s activities of daily living. Grade 3 hand-and-foot syndrome is defined as moist desquamation, ulceration, blistering or severe pain of the hands and/or feet and/or severe discomfort that causes the patient to be unable to work or perform activities of daily living. If grade 2 or 3 hand-and-foot syndrome occurs, administration of XELODA should be interrupted until the event resolves or decreases in intensity to grade 1. Following grade 3 hand-and-foot syndrome, subsequent doses of XELODA should be decreased [see Dosage and Administration (2.2)].5.8 HyperbilirubinemiaIn 875 patients with either metastatic breast or colorectal cancer who received at least one dose of XELODA 1250 mg/m2 twice daily as monotherapy for 2 weeks followed by a 1-week rest period, grade 3 (1.5-3 x ULN) hyperbilirubinemia occurred in 15.2% (n=133) of patients and grade 4 (>3 x ULN) hyperbilirubinemia occurredhepatic metastases at baseline, grade 3 or 4 hyperbilirubinemia occurred in 22.8% and 12.3%, respectively. Of the 167 patients with grade 3 or 4 hyperbilirubinemia, 18.6% (n=31) also had postbaseline elevations (grades 1 to 4, without elevations at baseline) in alkaline phosphatase and 27.5% (n=46) had postbaseline elevations in transaminases at any time (not necessarily concurrent). The majority of these patients, 64.5% (n=20) and 71.7% (n=33), had liver metastases at baseline. In addition, 57.5% (n=96) and 35.3% (n=59) of the 167 patients had elevations (grades 1 to 4) at both prebaseline and postbaseline in alkaline phosphatase or transaminases, respectively. Only 7.8% (n=13) and 3.0% (n=5) had grade 3 or 4 elevations in alkaline phosphatase or transaminases.In the 596 patients treated with XELODA as first-line therapy for metastatic colorectal cancer, the incidence of grade 3 or 4 hyperbilirubinemia was similar to the overall clinical trial safety database of XELODA monotherapy. The median time to onset for grade 3 or 4 hyperbilirubinemia in the colorectal cancer population was 64 days and median total bilirubin increased from 8 μm/L at baseline to 13 μm/L during treatment with XELODA. Of the 136 colorectal cancer patients with grade 3 or 4 hyperbilirubinemia, 49 patients had grade 3 or 4 hyperbilirubinemia as their last measured value, of which 46 had liver metastases at baseline.In 251 patients with metastatic breast cancer who received a combination of XELODA and docetaxel, grade 3 (1.5 to 3 x ULN) hyperbilirubinemia occurred in 7% (n=17) and grade 4 (>3 x ULN) hyperbilirubinemia occurred in 2% (n=5).If drug-related grade 3 to 4 elevations in bilirubin occur, administration of XELODA should be immediately interrupted until the hyperbilirubinemia decreases to ≤3.0 X ULN [see recommended dose modifications under Dosage and Administration (2.2)].5.9 HematologicIn 875 patients with either metastatic breast or colorectal cancer who received a dose of 1250 mg/m2 administered twice daily as monotherapy for 2 weeks followed by a 1-week rest period, 3.2%, 1.7%, and 2.4% of patients had grade 3 or 4 neutropenia, thrombocytopenia or decreases in hemoglobin, respectively. In 251 patients with metastatic breast cancer who received a dose of XELODA in combination with docetaxel, 68% had grade 3 or 4 neutropenia, 2.8% had grade 3 or 4 thrombocytopenia, and 9.6% had grade 3 or 4 anemia. Patients with baseline neutrophil counts of <1.5 x 109/L and/or thrombocyte counts of <100 x 109/L should not be treated with XELODA. If unscheduled laboratory assessments during a treatment cycle show grade 3 or 4 hematologic toxicity, treatment with XELODA should be interrupted.5.10 Geriatric PatientsPatients ≥80 years old may experience a greater incidence of grade 3 or 4 adverse reactions. In 875 patients with either metastatic breast or colorectal cancer who received XELODA monotherapy, 62% of the 21 patients ≥80 years of age treated with XELODA experienced a treatment-related grade 3 or 4 adverse event: diarrhea in 6 (28.6%), nausea in 3 (14.3%), hand-and-foot syndrome in 3 (14.3%), and vomiting in 2 (9.5%) patients. Among the 10 patients 70 years of age and greater (no patients were >80 years of age) treated with XELODA in combination with docetaxel, 30% (3 out of 10) of patients experienced grade 3 or 4 diarrhea and stomatitis, and 40% (4 out of 10) experienced grade 3 hand-and-foot syndrome.Among the 67 patients ≥60 years of age receiving XELODA in combination with docetaxel, the incidence of grade 3 or 4 treatment-related adverse reactions, treatment-related serious adverse reactions, withdrawals due to adverse reactions, treatment discontinuations due to adverse reactions and treatment discontinuations within the first two treatment cycles was higher than in the <60 years of age patient group.In 995 patients receiving XELODA as adjuvant therapy for Dukes’ C colon cancer after resection of the primary tumor, 41% of the 398 patients ≥65 years of age treated with XELODA experienced a treatment-related grade 3 or 4 adverse event: hand-and-foot syndrome in 75 (18.8%), diarrhea in 52 (13.1%), stomatitis in 12 (3.0%), neutropenia/granulocytopenia in 11 (2.8%), vomiting in 6 (1.5%), and nausea in 5 (1.3%) patients. Inpatients ≥65 years of age (all randomized population; capecitabine 188 patients, 5-FU/LV 208 patients) treated for Dukes’ C colon cancer after resection of the primary tumor, the hazard ratios for disease-free survival and overall survival for XELODA compared to 5-FU/LV were 1.01 (95% C.I. 0.80 – 1.27) and 1.04 (95% C.I. 0.79 – 1.37), respectively.5.11 Hepatic InsufficiencyPatients with mild to moderate hepatic dysfunction due to liver metastases should be carefully monitored when XELODA is administered. The effect of severe hepatic dysfunction on the disposition of XELODA is not known [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. 5.12 Combination With Other DrugsUse of XELODA in combination with irinotecan has not been adequately studied.6 ADVERSE REACTIONSBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may notreflect the rates observed in practice.6.1 Adjuvant Colon CancerTable 4 shows the adverse reactions occurring in ≥5% of patients from one phase 3 trial in patients with Dukes’ C colon cancer who received at least one dose of study medication and had at least one safety assessment. Atotal of 995 patients were treated with 1250 mg/m 2 twice a day of XELODA administered for 2 weeks followedby a 1-week rest period, and 974 patients were administered 5-FU and leucovorin (20 mg/m 2leucovorin IVfollowed by 425 mg/m 2IV bolus 5-FU on days 1-5 every 28 days). The median duration of treatment was 164 days for capecitabine-treated patients and 145 days for 5-FU/LV-treated patients. A total of 112 (11%) and 73 (7%) capecitabine and 5-FU/LV-treated patients, respectively, discontinued treatment because of adverse reactions. A total of 18 deaths due to all causes occurred either on study or within 28 days of receiving study drug: 8 (0.8%) patients randomized to XELODA and 10 (1.0%) randomized to 5-FU/LV.Table 5 shows grade 3/4 laboratory abnormalities occurring in ≥1% of patients from one phase 3 trial inpatients with Dukes’ C colon cancer who received at least one dose of study medication and had at least one safety assessment. Table 4Percent Incidence of Adverse Reactions Reportedin ≥5% of Patients Treated With XELODA or 5-FU/LV for Colon Cancer in the Adjuvant Setting (Safety Population)Adjuvant Treatment for Colon Cancer (N=1969)XELODA (N=995) 5-FU/LV (N=974)Body System/ Adverse EventAll Grades Grade 3/4 All Grades Grade 3/4GastrointestinalDisordersDiarrhea Nausea Stomatitis Vomiting Abdominal Pain Constipation Upper Abdominal Pain47 34 22 15 14 9 7 12 2 2 2 3 -<1 65 47 60 21 16 11 7 14 2 14 2 2 <1<1。
奥沙利铂卡培他滨方案1. 简介奥沙利铂卡培他滨是一种常用于治疗多种恶性肿瘤的化疗方案。
本文将介绍奥沙利铂卡培他滨方案的使用方法、副作用以及注意事项。
2. 使用方法2.1 药物组合奥沙利铂卡培他滨方案由奥沙利铂和卡培他滨两种药物组合而成。
2.2 给药途径奥沙利铂卡培他滨方案通常以静脉输注的形式给药。
2.3 用药周期奥沙利铂卡培他滨方案的用药周期根据患者的具体情况而定,一般为21天一个周期。
每个周期包括药物给药的日子和休息的日子。
2.4 治疗方案奥沙利铂卡培他滨方案的具体治疗方案需要根据患者的癌症类型、病情严重程度等因素来确定。
一般情况下,医生会根据病情给予不同剂量和次数的药物给药。
3. 副作用奥沙利铂卡培他滨方案可能会引起一些副作用,包括但不限于以下几种:•恶心和呕吐:这是最常见的副作用之一。
医生会根据需要给患者开具相应的抗恶心药物来缓解这些症状。
•脱发:一些患者在接受奥沙利铂卡培他滨方案治疗期间会出现脱发的情况。
这是由于药物对毛发的影响导致的,一般在治疗结束后会逐渐恢复。
•骨髓抑制:奥沙利铂卡培他滨方案可能会影响骨髓造血功能,导致贫血、白细胞减少等情况。
医生会定期进行血常规检查,以确保患者的血液指标在正常范围内。
4. 注意事项在接受奥沙利铂卡培他滨方案治疗期间,患者需要注意以下事项:•饮食调理:合理的饮食可以帮助患者更好地应对治疗期间的身体不适。
建议患者避免油腻、刺激性食物,多摄入富含营养的食物。
•休息调节:适当休息对于患者的身体恢复非常重要。
患者在治疗期间需要注意休息,避免过度劳累。
•定期复诊:患者需要按医生的要求定期复诊,进行相关的检查和评估,以监测治疗的效果和副作用情况。
5. 结论奥沙利铂卡培他滨方案是一种常用的化疗方案,可用于多种恶性肿瘤的治疗。
在接受治疗期间,患者需要密切关注副作用的出现,遵循医生的建议并注意个人的饮食和休息调节。
在治疗过程中,及时与医生沟通,定期复诊,以确保治疗的效果和患者的健康状态。
卡培他滨 (Capecitabine) 癌症的化疗药物卡培他滨 (Capecitabine)——癌症的化疗药物癌症是一种严重威胁人类健康和生命的疾病,对患者和家庭都造成了巨大的心理和经济负担。
随着医疗技术的不断进步,化疗作为治疗癌症的重要手段之一,为患者带来了希望。
卡培他滨 (Capecitabine)作为一种常用的化疗药物,在癌症治疗中发挥着重要的作用。
卡培他滨是一种口服抗癌药物,属于抑制胸苷酸合成酶的药物,其主要成分为卡培他滨。
作为一种眼下使用普遍的抗癌药物,卡培他滨已经被广泛应用于多种癌症的治疗当中。
首先,卡培他滨在乳腺癌的治疗中具有重要意义。
乳腺癌是世界各国女性常见的恶性肿瘤之一,卡培他滨作为一线治疗方案中的重要组成部分,已经获得了广泛的应用。
卡培他滨通过抑制胸苷酸合成酶,从而抑制乳腺癌细胞的增殖和扩散,起到了很好的治疗效果。
其次,卡培他滨在结直肠癌的治疗中也扮演着重要的角色。
结直肠癌是常见的消化系统肿瘤,发病率逐年上升。
卡培他滨通过抑制肿瘤细胞中胸苷酸合成酶的活性,从而抑制结直肠癌细胞的增殖和分裂,达到治疗效果。
临床研究显示,结直肠癌患者通过卡培他滨进行化疗,可以显著提高生存率和生存质量。
此外,卡培他滨还可以用于胃癌、食管癌、头颈部癌症等多种癌症的治疗。
它通过干扰肿瘤细胞的DNA合成和RNA转录,从而阻断癌细胞的增殖和转移。
卡培他滨的药代动力学也使其成为化疗的十分理想的药物,通过口服给药,能够更好的为患者提供临床治疗。
同时,由于卡培他滨具有足够的安全性和容忍性,患者在使用该药物期间通常不会出现明显的毒副作用,有利于患者的接受和治疗效果的提高。
总之,卡培他滨作为一种重要的癌症化疗药物,已经在临床实践中发挥了重要的作用。
它通过抑制胸苷酸合成酶,抑制癌细胞的生长和扩散,有效治疗乳腺癌、结直肠癌等多种癌症。
随着医疗技术的不断创新,相信卡培他滨会在未来的癌症治疗中发挥更加重要的作用,为患者带来更多的康复希望。
卡培他滨说明书
请仔细阅读说明书并在医师指导下使用
警告!
对于同时服用卡培她滨和香豆素类衍生物抗凝药如华法令和苯丙香豆素的患者,应该频繁监测抗凝反应指标,如INR或凝血酶原时间,以调整抗凝剂的用量。
在合并用药期间,曾有凝血参数改变和/或出血,包括死亡的报告。
发生时间:在开始卡培她滨治疗后几天到几个月时间内,也可能在停止使用卡培她滨后1个月内观察到。
易感因素:年龄>60,诊断为癌症。
【药品名称】
通用名称:卡培她滨片
英文名称:Capecitabine Tablets
汉语拼音:Kapeitabin Pian
【成份】
本品主要成份为卡培她滨。
化学名称:5’-脱氧-5-氟-N-[(戊氧基)羰基]-胞(嘧啶核)苷。
分子式:C15H22FN3O6
分子量:359.35
【性状】
本品为薄膜衣片,除去包衣后显白色或类白色。
【适应症】
结肠癌辅助化疗:卡培她滨适用于Dukes’ C期、原发肿瘤根治术后、适于接受氟嘧啶类药物单独治疗的结肠癌患者的单药辅助治疗。
其治疗的无病生存期(DFS)不亚于5-氟尿嘧啶和甲酰四氢叶酸联合方案(5-FU/LV) 。
卡培她滨单药或与其它药物联合化疗均不能延长总生存期(OS),但已有试验数据表明在联合化疗方案中卡培她滨可较5-FU/LV改进无病生存期。
医师在开具处方使用卡培她滨单药对Dukes’ C期结肠癌进行辅助治疗时,可参考以上研究结果。
结直肠癌:卡培她滨单药或与奥沙利铂联合(XELOX)适用于转移性结直肠癌的一线治疗。
乳腺癌联合化疗:卡培她滨可与多西紫杉醇联合用于治疗含蒽环类药物方案化疗失败的转移性乳腺癌。
乳腺癌单药化疗:卡培她滨亦可单独用于治疗对紫杉醇及含蒽环类药物化疗方案均耐药或对紫杉醇耐药和不能再使用蒽环类药物治疗(例如已经接受了累积剂量400 mg/m2阿霉素或阿霉素同类物)的转移性乳腺癌患者。
耐药的定义为治疗期间疾病继续进展(有或无初始缓解),或完成含有蒽环类药物的辅助化疗后6个月内复发。
胃癌:卡培她滨适用于不能手术的晚期或者转移性胃癌的一线治疗。
【规格】0.5g
【用法用量】
卡培她滨的推荐剂量为1250mg/m2,每日2次口服(早晚各1次;等于每日总剂量2500mg/m2),治疗2周后停药1周,3周为一个疗程。
卡培她滨片剂应在餐后30分钟内用水吞服。
在与多西紫杉醇联合使用时,卡培她滨的推荐剂量为1250 mg/m2,每日2次,治疗2周后停药1周,与之联用的多西紫杉醇推荐剂量为75 mg/m2,每3周1次,静脉滴注1小时。
根据多西紫杉醇的说明书,在对接受卡培她滨和多西紫杉醇联合化疗的患者使用多西紫杉醇前,应常规应用一些化疗辅助药物。
与奥沙利铂联合使用时,在对患者给予奥沙利铂(剂量为130 mg/m2,静脉输注2小时)后的当天即可开始卡培她滨的治疗,剂量为1000 mg/m2,每日2次,治疗2周后停药1周。
有关奥沙利铂用药以及用药前给予预治疗药物的详细信息,参见奥沙利铂的药品说明书。
表1和表2分别介绍了卡培她滨的起始剂量为1250 mg/m2或1000 mg/m2时,标准剂量和降低剂量的计算方法(见剂量调整指南)。
当用于Dukes’ C期结肠癌患者的辅助治疗时,推荐治疗时间为6个月,即卡培她滨1250mg/m2,每日2次口服,治疗2周后停药1周,以3周为一个疗程,共计8个疗程(24周)。
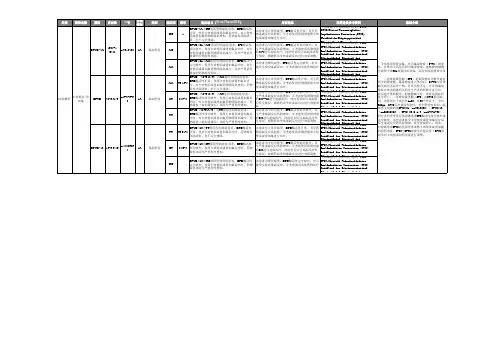
表1. 根据体表面积计算的卡培她滨标准剂量和降低后的剂量,
起始剂量1250mg/m2
*每日总剂量分为早晚各1次口服,早晚剂量相等
表2. 根据体表面积计算的卡培她滨标准剂量和降低后的剂量,
1000mg/m2
起始剂量
*每日总剂量分为早晚各1次口服,早晚剂量相等
剂量调整指南:
在使用中卡培她滨用药剂量可能需要调整,以达到适应患者个体化的需求。
使用中应密切监测不良反应,并根据需要调整剂量以使患者能够耐受治疗。
卡培她滨所致的不良反应可经过对症治疗、停药和调整剂量等方式处理。
药物一经减量,以后便不应再增加剂量。
当苯妥英和香豆素衍生物类抗凝剂类药物与卡培她滨合用时,可能需要减量(见【药物相互作用】:抗凝剂)。
发生不良反应时,卡培她滨的剂量调整方案可参照下表进行处理(见表3和表4)。
表3. 卡培她滨联合多西紫杉醇化疗时剂量调整方案。