XRD晶体结构分析 2
- 格式:ppt
- 大小:24.25 MB
- 文档页数:133


材料课堂——XRD常见问题详解(二),超实用!!展开全文衍射峰的强度和很多因素有关,比如样品的衍射能力,性质,还有仪器功率,测试方法,检测器的灵敏度等等。
XRD 衍射强度和峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关?样品中晶粒越小,衍射峰的峰高强度越来越低,但是峰越来越宽,实际上利用X 射线衍射峰的宽化对样品的结晶颗粒度分析就是根据这个原理的(Scherrer 公式)。
晶粒大小和颗粒大小有关系,但是其各自的含义是有区别的。
一颗晶粒也可能就是一颗颗粒,但是更可能的情况是晶粒抱到一起 , 二次聚集, 成为颗粒。
颗粒不是衍射的基本单位, 但是微小的颗粒能产生散射。
你磨的越细, 散射就越强.。
对于晶粒, 你磨过头了, 晶体结构被破坏了, 磨成非晶, 衍射能力就没有了。
磨得太狠的话,有些峰可能要消失了,而且相邻较近的衍射峰会由于宽化而相互叠加,最终会变成1 个或几个'鼓包'。
一般晶面间距大的峰受晶粒细化的影响会明显一些,因为 d 值大的晶面容易被破坏。
衍射强度变弱本质的原因是由于晶体颗粒变小,还是样品颗粒变小?强度除了和晶粒度有关外,还和晶粒的表面状态有关。
一般颗粒越细,其表面积越大,表面层结构的缺陷总是比较严重的。
结构缺陷将导致衍射强度降低和衍射峰宽化。
XRD 研究的应该是晶粒、晶体的问题,与晶体结构关联的问题,不是样品颗粒的大小问题,谢乐公式算的应该也是晶粒的大小。
样品颗粒的大小要用别的方法测定.,例如光散射、X 射线散射、电镜等。
细针状微晶粉末样品做 XRD 重复性很差。
(制作粉末衍射样品片)怎么可以避免择优取向?择优取向是很难避免的,只能尽力减少他的影响。
首先,你要讲样品磨得尽量细(但要适度,要注意样品的晶体结构不要因研磨过度而受到损坏);不要在光滑的玻璃板上大力压紧(压样时可以在玻璃板上衬一张粗糙的纸张),样品成形尽可能松一些;制样过程中也可以掺一些玻璃粉,或加一些胶钝化一下样品的棱角。


XRD,以及晶体结构的相关基础知识(ZZ)Theory 2009-10-25 17:55:42 阅读355 评论0 字号:大中小做XRD有什么用途啊,能看出其纯度?还是能看出其中含有某种官能团?X射线照射到物质上将产生散射。
晶态物质对X射线产生的相干散射表现为衍射现象,即入射光束出射时光束没有被发散但方向被改变了而其波长保持不变的现象,这是晶态物质特有的现象。
绝大多数固态物质都是晶态或微晶态或准晶态物质,都能产生X射线衍射。
晶体微观结构的特征是具有周期性的长程的有序结构。
晶体的X射线衍射图是晶体微观结构立体场景的一种物理变换,包含了晶体结构的全部信息。
用少量固体粉末或小块样品便可得到其X射线衍射图。
XRD(X射线衍射)是目前研究晶体结构(如原子或离子及其基团的种类和位置分布,晶胞形状和大小等)最有力的方法。
XRD 特别适用于晶态物质的物相分析。
晶态物质组成元素或基团如不相同或其结构有差异,它们的衍射谱图在衍射峰数目、角度位置、相对强度次序以至衍射峰的形状上就显现出差异。
因此,通过样品的X射线衍射图与已知的晶态物质的X射线衍射谱图的对比分析便可以完成样品物相组成和结构的定性鉴定;通过对样品衍射强度数据的分析计算,可以完成样品物相组成的定量分析;XRD还可以测定材料中晶粒的大小或其排布取向(材料的织构)...等等,应用面十分普遍、广泛。
目前XRD主要适用于无机物,对于有机物应用较少。
关于XRD的应用,在[技术资料]栏目下有介绍更详细的文章,不妨再深入看看。
如何由XRD图谱确定所做的样品是准晶结构?XRD图谱中非晶、准晶和晶体的结构怎么严格区分?三者并无严格明晰的分界。
在衍射仪获得的XRD图谱上,如果样品是较好的"晶态"物质,图谱的特征是有若干或许多个一般是彼此独立的很窄的"尖峰"(其半高度处的2θ宽度在0.1°~0.2°左右,这一宽度可以视为由实验条件决定的晶体衍射峰的"最小宽度")。

XRD图谱分析最终版XRD图谱分析解析X射线衍射谱图中,d是晶体晶格中相邻两个晶面的面间距,一般以埃为单位。
晶体的空间结构可以用三轴坐标系表示,也可以用四轴定向表示,尤其是三方、六方晶系用四轴定向表示有其独到的便利。
在三轴定向中,在不同晶向,相邻两个晶面间的晶面间距都可以用d表示。
d的脚标用其所描述的正点阵或倒易点阵的相应晶面指标(hkl)表示。
如:d(100),d(020),d(002),等。
在研究石墨状微晶、多晶石墨或碳纳米管、碳纤维等类石墨结构等材料的X射线衍射测定中,发现石墨、类石墨晶体结构的X射线衍射谱的峰并不多。
常用d002代表石墨状微晶的平均层层间距;用Lc表示微晶层面沿c轴方向(有时刚好也就是002晶面指数,可以使用 002 峰参数进行计算)的堆积厚度;用La表示沿a轴方向的微晶宽度或直径等面间距,使用100峰或110峰(要视具体晶体而定)进行计算。
对于晶体或部分晶体样品的X射线衍射谱解析讨论中,三个晶向上的晶面间距都可以用d表示之,而不论它是否经过拉伸或加温处理而改变晶格结构与否,它都是作为一个一个晶体样品、晶体对象存在的。
但如果在一个系列中,主要研究点是通过加力、加温而使晶体发生变化,再用d表示三轴方向上的面间距就不如使用另一些字母以显示其特点而避免与常规面间距d混淆,La,Lc就是这样应运而生了。
X射线衍射分析,是以布拉格定律(公式)为基础的。
布拉格公式: 2d sinθ=nλ,式中λ为X射线的波长(Cuka 波长为0.15406nm,Cuka1 波长为0.15418nm。
)n为任何正整数,并相应称为n级衍射。
θ是掠射角(也称布拉格角,是入射角的余角),2θ才是衍射角。
当X射线以掠射角θ入射到某一点阵平面间距为d的原子面上时,在符合上式的条件下,将在反射方向上得到因叠加而加强的衍射线。
布拉格定律简洁直观地表达了衍射所必须满足的条件。
当X射线波长λ已知时(选用固定波长的特征X射线),采用细粉末或细粒多晶体的线状样品,可从一堆任意取向的晶体中,从每一个θ角符合布拉格条件的反射面得到反射。

xrd分析2篇第一篇:X射线衍射(XRD)分析X射线衍射(XRD)是一种常见的非破坏性分析技术,可用于确定晶体结构及其组成成分。
XRD分析方法的原理基于晶体中的原子或离子对于X射线的反射。
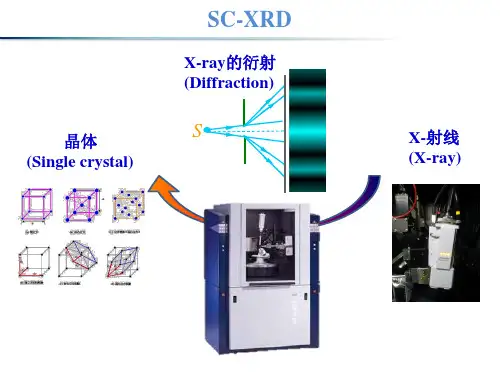
在XRD实验中,样品被置于一个被称为X射线衍射仪的仪器中。
该仪器通过一个小孔射出高能X射线束,该束被定向到通过样品的X射线探测器上。
当X射线进入样品时,它们会与样品中的原子或离子相互作用,并发生散射。
这种散射会以特定的角度返回到探测器中,并产生X射线衍射图谱。
这个图谱显示出了在特定角度处的衍射强度。
每个晶体结构对应的衍射角度和强度都是唯一的。
因此,通过比较实验结果与已知晶体结构库中的标准谱图,可以确定样品的晶体结构和成分。
XRD分析方法在许多领域都有广泛的应用,例如材料科学、地质学、生物学和环境科学等。
它可以用于确定化合物、制备材料和表征材料的性质。
由于XRD是一种非破坏性分析方法,因此它可以在不破坏样品的情况下提供与样品相关的宝贵信息。
总之,XRD分析方法是一种可靠的技术,具有许多广泛的应用。
通过使用XRD,可以轻松确定晶体结构和成分,并帮助科学家更好地理解材料和化合物的性质。
第二篇:X射线衍射(XRD)分析的优势和限制X射线衍射(XRD)是确定晶体结构和成分的常见技术。
尽管这种分析有许多优点,但它也存在一些局限性。
首先,XRD分析具有高分辨率和高灵敏度。
它可以确定极小的结构细节,并区分具有类似晶体结构的样品。
此外,由于XRD是一种非破坏性分析方法,因此可以在不破坏样品的情况下获得所需信息。
然而,XRD分析也有一些限制。
首先,由于样品必须是晶体形式,因此非晶体或无序材料将无法进行XRD分析。
其次,XRD只适用于固体材料,因此液态或气态的样品需要转化为固体形式才能进行分析。
此外,XRD分析也会受到诸如样品晶面朝向、晶粒大小、杂质含量、衍射峰形等因素的影响。
这些因素可能会造成误判或分析结果的不确定性。
总之,XRD分析是一种可靠的技术,但也具有局限性。




XRD及其晶体结构的相关知识X射线荧光衍射:利用初级X射线光子或其他微观离子激发待测物质中的原子,使之产生荧光(次级X射线)而进行物质成分分析和化学态研究的方法。
按激发、色散和探测方法的不同,分为X射线光谱法(波长色散)和X射线能谱法(能量色散)。
当原子受到X射线光子(原级X射线)或其他微观粒子的激发使原子内层电子电离而出现空位,原子内层电子重新配位,较外层的电子跃迁到内层电子空位,并同时放射出次级X射线光子,此即X射线荧光。
较外层电子跃迁到内层电子空位所释放的能量等于两电子能级的能量差,因此,X射线荧光的波长对不同元素是特征的。
根据色散方式不同,X射线荧光分析仪相应分为X射线荧光光谱仪(波长色散)和X射线荧光能谱仪(能量色散)。
X射线荧光光谱仪主要由激发、色散、探测、记录及数据处理等单元组成。
激发单元的作用是产生初级X射线。
它由高压发生器和X光管组成。
后者功率较大,用水和油同时冷却。
色散单元的作用是分出想要波长的X射线。
它由样品室、狭缝、测角仪、分析晶体等部分组成。
通过测角器以1∶2速度转动分析晶体和探测器,可在不同的布拉格角位置上测得不同波长的X射线而作元素的定性分析。
探测器的作用是将X射线光子能量转化为电能,常用的有盖格计数管、正比计数管、闪烁计数管、半导体探测器等。
记录单元由放大器、脉冲幅度分析器、显示部分组成。
通过定标器的脉冲分析信号可以直接输入计算机,进行联机处理而得到被测元素的含量。
X射线荧光能谱仪没有复杂的分光系统,结构简单。
X射线激发源可用X射线发生器,也可用放射性同位素。
能量色散用脉冲幅度分析器。
探测器和记录等与X射线荧光光谱仪相同。
X射线荧光光谱仪和X射线荧光能谱仪各有优缺点。
前者分辨率高,对轻、重元素测定的适应性广。
对高低含量的元素测定灵敏度均能满足要求。
后者的X射线探测的几何效率可提高2~3数量级,灵敏度高。
可以对能量范围很宽的X 射线同时进行能量分辨(定性分析)和定量测定。
xx大学材料科学实验实验报告实验名称:XRD 测试晶体结构与精修辅导员意见:材料系专业班第实验小组作者学号实验日期年月日成绩:辅导员签名一、实验目的1.学习X-射线衍射物相定量分析的方法和步骤2.了解X-射线衍射精确测定晶胞参数的方法3.掌握Pullprof 晶体结构的精修二、实验原理:X射线在晶体中的衍射光波经过狭缝将产生衍射现象。
狭缝的大小必须与光波的波长同数量级或更小。
由图4-4a可知,当入射X射线与晶面相交θ角时,假定晶面就是镜面(即布拉格面,入射角与出射角相等),那末容易看出,图中两条射线1和2的光程差是AC+ DC,即2dsin θ。
当它为波长的整数倍时(假定入射光为单色的,只有一种波长)2d sin θ = nλ ,n =1,2,布拉格(Bragg)公式在θ 方向射出的X 射线即得到衍射加强。
根据布拉格公式,即可以利用已知的晶体(d 已知)通过测θ角来研究未知X射线的波长;也可以利用已知X射线(λ 已知)来测量未知晶体的晶面间距。
三、实验步骤1. 样品制备2. XRD 测试样品3. 学生自带笔记电脑,安装Fullprof 和UltraEdit 程序(老师给)4. 结构精修5. 结构输出及分析四、晶体结构测试与精修时应注意事项1、晶体的各向异性温度因子是如何定义的?答:晶体中的原子普遍存在热运动,这种运动在绝对零度时也未必停止。
通常所谓的原子坐标是指它们在不断振动中的平衡位置。
随着温度的升高,其振动的振幅增大。
这种振动的存在增大了原子散射波的位相差,影响了原子的散射能力,即衍射强度。
在晶体中,特别是对称性低的晶体,原子各个方向的环境并不相同,因此严格的说不同方向的振幅是不等的,由此引入了各向异性温度因子。
2、在进行Rietveld 结构精修时,是否该对温度因子进行约束?如何约束以及约束范围?答:由于温度因子是随着衍射角的增加而对强度的影响增大,所以,如果要精修温度因子,就一定要收集高角度的数据。
X射线衍射法测定晶体结构一、实验目的1.了解X射线衍射的基本原理及仪器装置;2.理解粉末衍射的XRD分析测试方法,并应用XRD 数据进行物相分析。
二、实验原理X射线衍射分析(X-ray diffraction,简称XRD),是利用晶体形成的X射线衍射,对物质进行内部原子在空间分布状况的结构分析方法。
将具有一定波长的X射线照射到结晶性物质上时,X射线因在结晶内遇到规则排列的原子或离子而发生散射,散射的X射线在某些方向上相位得到加强,从而显示与结晶结构相对应的特有的衍射现象。
X射线衍射方法具有不损伤样品、无污染、快捷、测量精度高、能得到有关晶体完整性的大量信息等。
晶体对X射线的衍射,归根结底是晶体中原子的电子对X射线的相干散射。
当X射线电磁波作用于电子后,电子在其电场力作用下,将随着X射线的电场一起震动,成为一个发射电磁波的波源,其震动频率与X射线频率相同。
一个单原子能使一束X射线向空间所有方向散射。
但数目很大的原子在三维空间里呈点阵形式排列成晶体时,由于散射波之间的互相干涉,所以只有在某些方向上才产生衍射。
衍射方向取决于晶体内部结构周期重复的方式和晶体安置的方位。
测定晶体的衍射方向,可以求得晶胞的大小和形状。
联系衍射方向和晶胞大小形状间关系的方程有两个:Laue(劳)方程和Bragg(布拉格)方程。
前者以直线点阵为出发点,后者以平面点阵为出发点,这两个方程是等效的,可以互推。
晶体的X射线衍射图像实质上是晶体微观结构的一种精细复杂的变换,每种晶体的结构与其X射线衍射图之间都有着一一对应的关系,其特征X 射线衍射图谱不会因为它种物质混聚在一起而产生变化,这就是X射线衍射物相分析方法的依据。
制备各种标准单相物质的衍射花样并使之规范化,将待分析物质的衍射花样与之对照,从而确定物质的组成相,就成为物相定性分析的基本方法。
三、仪器设备本实验使用的仪器是Rigaku Ultima X射线衍射仪。
主要由冷却循环水系统、X射线衍射仪和计算机控制处理系统三部分组成。