使用CRISPR-CAS系统构建可遗传的基因敲除小鼠和大鼠
- 格式:doc
- 大小:25.50 KB
- 文档页数:3

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言基因编辑技术近年来在生物学和医学领域取得了巨大的突破,其中CRISPR-Cas9系统因其高效、精确的特性,已成为基因编辑的主要工具之一。
本文旨在探讨利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的方法和过程,为基因编辑技术的进一步应用提供参考。
二、DUSP9基因与小鼠胚胎干细胞DUSP9基因是一种重要的蛋白磷酸酶基因,其编码的蛋白在细胞信号传导过程中具有重要作用。
小鼠胚胎干细胞(Mouse Embryonic Stem Cells, mESCs)是研究发育生物学和基因编辑的重要工具。
通过构建DUSP9基因敲除的小鼠胚胎干细胞系,可以研究DUSP9基因在细胞发育、分化以及疾病发生过程中的作用。
三、CRISPR-Cas9系统简介CRISPR-Cas9系统是一种基于细菌免疫系统的基因编辑技术,其原理是通过将特定的RNA与Cas9蛋白结合,形成复合物,然后识别并切割DNA序列,从而实现对基因的敲除或修改。
CRISPR-Cas9系统具有精确度高、效率高、成本低等优点,是现代基因编辑的主要手段。
四、构建DUSP9基因敲除小鼠胚胎干细胞系的步骤1. 载体构建:设计并合成针对DUSP9基因的特异性gRNA 序列,并将其与Cas9蛋白的表达载体一起构建成CRISPR-Cas9表达系统。
2. 细胞培养与转染:将小鼠胚胎干细胞培养至适当状态,然后利用转染技术将CRISPR-Cas9表达系统导入细胞中。
3. 基因编辑:通过CRISPR-Cas9系统识别并切割DUSP9基因的DNA序列,实现DUSP9基因的敲除。
4. 克隆筛选与鉴定:筛选并培养获得成功的DUSP9基因敲除小鼠胚胎干细胞克隆,通过PCR、测序等方法鉴定敲除效果。
5. 细胞系建立与保存:将成功构建的DUSP9基因敲除小鼠胚胎干细胞系进行保存与扩大培养,以备后续研究使用。

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言随着基因编辑技术的发展,CRISPR-Cas9系统已成为一种强大的工具,用于在生物医学研究中精确地编辑基因组。
DUSP9基因作为一种重要的基因,其功能在多种生物学过程中起着关键作用。
因此,构建DUSP9基因敲除小鼠胚胎干细胞系,对于研究DUSP9基因的功能及其在疾病发生发展中的作用具有重要意义。
本文旨在详细介绍利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的过程。
二、材料与方法1. 材料小鼠胚胎干细胞(mESCs)、CRISPR-Cas9系统、相关基因编辑工具、培养基、生长因子等。
2. 方法(1)设计CRISPR-Cas9系统:根据DUSP9基因的序列信息,设计合适的CRISPR-Cas9系统,包括sgRNA和Cas9蛋白。
(2)制备mESCs细胞:培养mESCs细胞至合适的状态,以便进行基因编辑。
(3)转染与编辑:将CRISPR-Cas9系统转染至mESCs细胞中,利用Cas9蛋白对DUSP9基因进行切割。
(4)筛选与鉴定:通过PCR、Western blot、qRT-PCR等方法,筛选出成功敲除DUSP9基因的mESCs细胞,并进行鉴定。
三、实验过程1. 设计并构建CRISPR-Cas9系统,选择合适的sgRNA序列和Cas9蛋白表达载体。
2. 培养mESCs细胞至合适的状态,进行转染。
3. 观察转染后的细胞生长情况,确保Cas9蛋白的表达。
4. 利用PCR、Western blot、qRT-PCR等方法筛选出成功敲除DUSP9基因的mESCs细胞。
5. 对筛选出的细胞进行扩增培养,并保存于液氮中备用。
四、结果与讨论1. 结果(1)成功构建了CRISPR-Cas9系统,并将其转染至mESCs 细胞中。
(2)成功筛选出敲除DUSP9基因的mESCs细胞,并通过PCR、Western blot、qRT-PCR等方法进行了鉴定。

《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言随着基因编辑技术的发展,CRISPR-Cas9系统已成为现代生物医学研究中常用的基因编辑工具之一。
它为科研人员提供了强大的基因敲除、插入或突变的能力,在多种模型动物制备及疾病研究领域具有广泛的应用前景。
本文旨在介绍利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系的过程,为相关研究提供技术参考。
二、材料与方法1. 材料(1) CRISPR-Cas9系统相关组件(包括Cas9蛋白、sgRNA 等);(2) 小鼠胚胎干细胞系;(3) DUSP9基因特异性敲除载体;(4) 培养基、试剂及其他实验耗材。
2. 方法(1) 设计并构建DUSP9基因敲除载体;(2) 准备小鼠胚胎干细胞系并进行细胞培养;(3) 将DUSP9基因敲除载体与胚胎干细胞共培养,实现基因编辑;(4) 筛选并扩增成功敲除DUSP9基因的胚胎干细胞;(5) 对敲除细胞进行鉴定及保存。
三、实验过程1. DUSP9基因敲除载体的构建根据DUSP9基因序列,设计并合成sgRNA序列,构建DUSP9基因敲除载体。
通过PCR扩增获得目的片段,将其克隆至载体中,构建成功后的载体通过测序验证其准确性。
2. 小鼠胚胎干细胞的培养与准备将小鼠胚胎干细胞置于适宜的培养条件下进行培养,待细胞生长至适宜状态时进行后续实验。
3. 基因编辑及筛选将DUSP9基因敲除载体与小鼠胚胎干细胞共培养,通过CRISPR-Cas9系统实现DUSP9基因的敲除。
随后,通过PCR、测序等方法筛选出成功敲除DUSP9基因的胚胎干细胞。
4. 鉴定与保存对筛选出的成功敲除DUSP9基因的胚胎干细胞进行鉴定,包括细胞形态观察、生长曲线绘制、基因型鉴定等。
将鉴定合格的细胞进行保存,以备后续实验使用。
四、结果与讨论1. 结果通过CRISPR-Cas9系统成功构建了DUSP9基因敲除小鼠胚胎干细胞系,并筛选出成功敲除DUSP9基因的细胞。

38基于CRISPR/Cas9技术的TRPS1基因敲除小鼠模型的构建李腾雁,刘文杰,赵宏,蔡建强*(国家癌症中心/ 国家肿瘤临床医学研究中心/ 中国医学科学院北京协和医学院肿瘤医院肝胆外科,北京 100021)李腾雁 博士研究生中国医学科学院北京协和医学院肿瘤医院肝胆外科目的:基于CRISPR/Cas9技术构建敲除TRPS1基因的杂合子小鼠,并进行鉴定。
方法: C57BL/6N小鼠自行交配后,使用Cas9/sgRNA注射受精卵的方法构建基因敲除小鼠,对可遗传的小鼠基因型进行鼠尾检测,TRPS1杂合子敲除小鼠分别与野生型小鼠交配,获得具有稳定基因型的小鼠。
结果:本实验通过使用Cas9/sgRNA注射受精卵的方法,所有繁殖小鼠经鼠尾基因型鉴定,证实成功构建了18只TRPS1基因敲除的杂合子小鼠。
结论:基于CRISPR/Cas9技术成功构建了敲除TRPS1基因的杂合子小鼠。
关键词:CRISPR/Cas9;TRPS1;结直肠癌;基因敲除小鼠摘要基金支持:国家自然科学基金(81672461) ;国家自然科学基金(81972311) ;深圳市“医疗卫生三名工程”(SZSM202011010)首都卫生发展科研专项项目(2018-1-4021);中国医学科学院医学与健康科技创新工程(2016-I2M-1-001,2017-12M-4-002) *通信作者:蔡建强************************Generation of TRPS1 knockout mice by CRISPR/Cas9-mediated gene targetingAbstractObjectives: This study aimed to construct and identify heterozygous mice knocked out of TRPS1 gene based on CRISPR/ Cas9 technology.Methods: After self-mating of C57BL/6N mice, TRPS1 knockout mice were constructed by injecting fertilized eggs with Cas9/sgRNA, and the mouse genotypes of heritable mice were detected by tail. TRPS1 heterozygous knockout mice were mated with wild-type mice to obtain mice with stable genotypes.Results: In this experiment, the fertilized eggs were injected with cas9 / sgRNA, all breeding mice were identified by tail genotype, 18 TRPS1 knockout heterozygous mice were successfully constructed.Conclusion: In this study, we successfully constructed TRPS1 knockout heterozygous mice based on CRISPR / cas9 technology, which provided a research platform for further research on the role of TRPS1 in the occurrence, development and possible liver metastasis of colorectal cancer at the animal level.Keywords: CRISPR/Cas9; TRPS1; Colorectal cancer; Gene knockout mouseLi Tengyan, Liu Wenjie, Zhao Hong, Cai Jianqiang*(National Department of Hepatobiliary Surgery, National Cancer Center/National Clinical Research Center for Cancer/ Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China)我国结直肠癌(colorectal cancer,CRC)的发病率和死亡率均保持上升趋势。
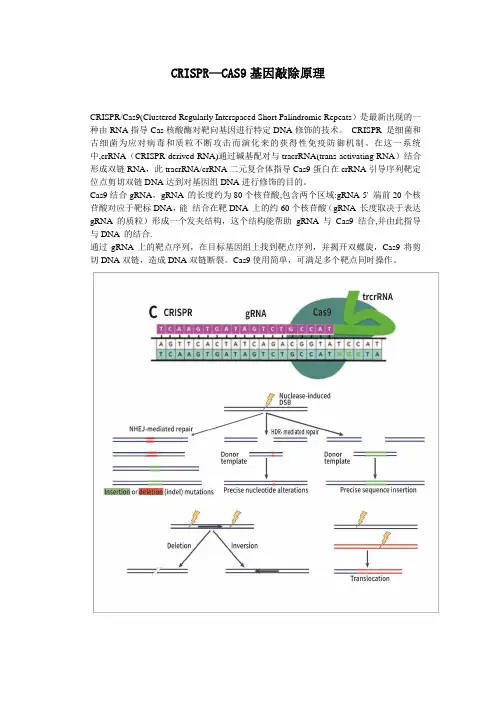
CRISPR—CAS9基因敲除原理
CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats)是最新出现的一种由RNA指导Cas核酸酶对靶向基因进行特定DNA修饰的技术。
CRISPR 是细菌和古细菌为应对病毒和质粒不断攻击而演化来的获得性免疫防御机制。
在这一系统中,crRNA(CRISPR-derived RNA)通过碱基配对与tracrRNA(trans-activating RNA)结合形成双链RNA,此tracrRNA/crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶定位点剪切双链DNA达到对基因组DNA进行修饰的目的。
Cas9结合gRNA,gRNA 的长度约为80个核苷酸,包含两个区域:gRNA 5' 端前20个核苷酸对应于靶标DNA,能结合在靶DNA 上的约60个核苷酸(gRNA 长度取决于表达gRNA 的质粒)形成一个发夹结构,这个结构能帮助gRNA 与Cas9结合,并由此指导与DNA 的结合.
通过gRNA上的靶点序列,在目标基因组上找到靶点序列,并揭开双螺旋,Cas9将剪切DNA双链,造成DNA双链断裂。
Cas9使用简单,可满足多个靶点同时操作。
Insertion /deletion NHEJ HDR
gRNA
Cas9
Donor vector
基因敲除小鼠流程:。

使用 CRISPR-Cas9 创建转基因小鼠的方案虽然近年来已经开发了几种基因组编辑工具,包括锌指结构和 TALENs(转录激活物样效应物核酸酶),但没有一种能像CRISPR/Cas9系统那样高效,该系统由一个RNA引导的DNA内切酶 (Cas9) 和对应的引导RNA(CRISPR) 组成。
利用该系统,研究人员能够实现一步敲除多个基因的等位基因的突变小鼠1。
只需两三周的时间,即可创造出子携带条件性等位基因和报告基因的小鼠2,并且该方案。
特别要注意的是,该过程不需要创建修改的小鼠ES细胞过程,该过程有时会十分困难3。
随着 Cas9 敲入和敲除小鼠的发展,预计越来越多的实验室将选择 CRISPR/Cas9 系统来生成转基因小鼠模型。
使用CRISPR-Cas9创建转基因小鼠的方案动物学研究。
2016 年 7 月 18 日;37(4): 205–213.利用 CRISPR/Cas9 和单倍体胚胎干细胞系统产生基因修饰的小鼠。
图 1.在小鼠胚胎上使用 CRISPR/Cas9 基因组编辑创建转基因小鼠的示意图。
通过共注射 Cas9 mRNA 和向指导 RNA,多个基因靶标可以在小鼠胚胎中一次敲除。
(改编自Yang H,Wang H 和 Jaenisch R. Nat Protoc。
2014 年 8 月;9(8):1956-68.)Sigma-Aldrich 是为基因组编辑提供工具和定制服务的领导者,包括 ZFN 和CRISPR/cas9。
默克还提供了广泛的小鼠胚胎验证培养基和试剂组合,用于储存、转移和扩增用于在EmbryoMAX™名下创建转基因小鼠模型的小鼠胚胎。
浏览所有的基因组编辑产品浏览所有经小鼠胚胎验证的试剂小鼠胚胎和ES细胞培养基小鼠ES细胞培养基实验方案和过程成功的小鼠模型项目的提示1.了解实验目的并开展研究。
生成正确的小鼠需要完全理解被测试依据的假设。
例如,研究者可能希望验证这样的假设:突变肝脏中的转运蛋白可能会减轻特定药物的肝毒性作用。

基于CRISPR Cas9技术基因敲除小鼠(Cas9-KO)的制作方法一、CRISPR/Cas9靶向基因敲除小鼠制作的基本技术原理:通过CRISPR/Cas9基因敲除技术,crRNA通过碱基配对与tracrRNA(trans-activating RNA)结合,形成双链RNA。
这一tracrRNA:crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶标的特定位点剪切双链DNA。
在与crRNA引导序列互补的位点,Cas9蛋白的HNH核酸酶结构域剪切互补链而Cas9 RuvC-like 结构域剪切非互补链,实现敲除目的基因的功能,制备基因敲除小鼠模型。
二、具体步骤如下:一)模型制作策略制作:利用生物信息学手段(NCBI&IMPC&MGI),分别仔细分析目的基因敲除后小鼠的生存能力及繁育能力,并结合邻近基因的影响,最终选择合适的敲除区域进行敲除方案的设计,出具相应的制作策略。
二)载体的设计和构建:使用麻省理工学院的CRISPR Design工具(/),依据中靶Score的高低及脱靶Score的高低设计一对长度为20bp的针对靶标DNA的寡聚核苷酸链序列用于制备sgRNA,并在该靶区域设计引物用于后续阳性小鼠的基因鉴定。
1、制备sgRNA的实验方法步骤:1)线性化pUC57-GDNA-T7载体中提pUC57-GDNA-T7载体,用BsaI线性化过夜。
胶回收保存备用。
2)引物退火及加磷酸将上下游引物(干粉)稀释,再进行引物退火及加磷酸。
3)连接&阳性菌落筛选取步骤二中的加磷酸产物与线性化载体pUC57-GDNA-T7进行连接,该连接反应在干式恒温器中进行。
对连接产物进行转化,涂板,37°C培养箱过夜培养。
再用PCR&测序的方法筛选阳性克隆,再将测序正确的克隆进行甘油菌保种,-80°C保存备用4)制备转录模板以构建好的sgRNA载体为模板进行PCR扩增,将PCR产物切胶回收,回收产物离心后倒掉上清留DNA沉淀,再溶解DNA。

《基于CRISPR-Cas9技术构建KDM2A基因敲除细胞系及S100A9基因敲除小鼠》基于CRISPR-Cas9技术构建KDM2A基因敲除细胞系及S100A9基因敲除小鼠一、引言近年来,基因编辑技术在生物学及医学研究领域取得了重大突破。
其中,CRISPR/Cas9技术以其高效、精确的基因编辑能力,在构建基因敲除模型、研究基因功能以及疾病机制等方面发挥了重要作用。
本文旨在介绍基于CRISPR/Cas9技术构建KDM2A基因敲除细胞系及S100A9基因敲除小鼠的实验过程、结果及分析。
二、材料与方法1. 材料(1)细胞系:选择适合进行基因编辑的细胞系。
(2)小鼠:选择适合的实验用小鼠。
(3)CRISPR/Cas9系统:包括Cas9蛋白、sgRNA等。
(4)实验试剂与设备:包括细胞培养试剂、PCR仪、显微镜等。
2. 方法(1)设计并合成针对KDM2A和S100A9基因的sgRNA。
(2)构建CRISPR/Cas9表达载体。
(3)将表达载体转染至细胞系或小鼠胚胎中。
(4)筛选并扩增基因敲除细胞系或小鼠。
(5)通过PCR、 Western blot等方法验证基因敲除效果。
三、实验结果1. KDM2A基因敲除细胞系的构建与验证(1)成功构建了针对KDM2A基因的CRISPR/Cas9表达载体,并转染至细胞系中。
(2)通过PCR和Western blot等方法,验证了KDM2A基因的成功敲除。
(3)KDM2A基因敲除细胞系在体外培养过程中表现出特定的生物学特性改变。
2. S100A9基因敲除小鼠的构建与验证(1)将CRISPR/Cas9表达载体注射至小鼠胚胎中,成功获得了S100A9基因敲除小鼠。
(2)通过PCR等方法,验证了S100A9基因的成功敲除。
(3)S100A9基因敲除小鼠在生长过程中表现出特定的表型变化。
四、讨论与分析1. KDM2A基因敲除细胞系的分析KDM2A是一种组蛋白去甲基酶,参与调控基因表达和细胞周期等过程。

第12卷第6期2021年3月Vol.12Mar.2021黑龙江科学HEILONGJIANG SCIENCE利用CRISPR/Cas9技术构建大鼠CCKAR敲除载体刘惊涛,李知宗,林显光,陈恒玲(中南民族大学生物医学工程学院/脑认知国家民委重点实验室/医学信息分析及肿瘤诊疗湖北省重点实验室,武汉430074)摘要:CCKAR在许多疾病的发病机理中具有重要作用。
为研究其分子机制,选择利用CRISPR/Cas9基因编辑体系,构建K0载体。
根据CCKAR基因序列设计敲除靶点,通过质粒单酶切、Oligo退火、连接,成功构建了CCKAR的敲除载体。
该质粒的成功构建可为将来CCKAR基因的功能研究提供重要的基础。
关键词:CRISPR/Cas9;CCKAR;敲除载体中图分类号:R361+1文献标志码:A文章编号:1674-8646(2021)06-0016-03Construction of Mice CCKAR Knockout Carrier by CRISPR/Cas9TechnologyLiu Jingtao,Li Zhizong,Lin Xianguang,Chen Hengling(College of Biomedical Engineering,South-Central University for Nationalities/Key Laboratory of Brain Cognition, State Ethnic Affairs Commission/Hubei Key Laboratory of Medical Information Analysisand Tumor Diagnosis and Treatment,Wuhan430074,China)Abstract:CCKAR has important function in pathogenesis of many diseases.In order to research the molecular mechanism,the research uses CRISPR/Cas9gene compiling system,and constructs KO carrier.According to CCKAR gene sequence design,target spot is knocked out.Through plasmid single digestion,Oligo anneal and connection,theknockout carrier of CCKAR is successfully constructed.The basis for future CCKAR genetic function research.Key words:CRISPR/Cas9;CCKAR;Knockout carrier1胆囊收缩素A受体与CRISPR/Cas9系统工作原理1.1胆囊收缩素A受体胆囊收缩素(Cholecystokinin,CCK)由肠I细胞分泌,CCK可通过内分泌、旁分泌、自分泌和神经分泌等多种形式调节多种细胞功能⑴。

《利用CRISPR-Cas9技术构建FGF21基因敲除小鼠及其对毛发生长的作用研究》篇一一、引言随着基因编辑技术的发展,CRISPR-Cas9技术已成为现代生物学研究的重要工具。
FGF21基因作为生长因子家族的一员,在多种生理过程中发挥着重要作用。
本研究旨在利用CRISPR-Cas9技术构建FGF21基因敲除小鼠模型,并探讨其对毛发生长的作用。
二、材料与方法1. 实验材料(1)实验动物:野生型小鼠;(2)基因编辑工具:CRISPR-Cas9系统;(3)实验试剂与设备:PCR仪、显微操作仪、荧光显微镜等。
2. 方法(1)FGF21基因敲除小鼠模型的构建:设计并合成针对FGF21基因的CRISPR-Cas9系统,通过显微注射技术将该系统注入小鼠受精卵中,获得基因敲除小鼠;(2)毛发生长观察:定期观察并记录野生型小鼠与FGF21基因敲除小鼠的毛发生长情况;(3)数据分析:采用统计学方法对实验数据进行处理与分析。
三、实验结果1. FGF21基因敲除小鼠模型的构建成功通过CRISPR-Cas9技术成功构建了FGF21基因敲除小鼠模型,经PCR及测序验证,确认了基因敲除的有效性。
2. 毛发生长观察结果(1)野生型小鼠毛发生长情况:野生型小鼠毛发生长正常,毛发密度、色泽及长度均无明显异常;(2)FGF21基因敲除小鼠毛发生长情况:FGF21基因敲除小鼠出现毛发稀疏、色泽暗淡、长度较短等现象。
3. 数据分析结果通过统计学方法对实验数据进行处理与分析,发现FGF21基因敲除小鼠的毛发生长情况与野生型小鼠存在显著差异(P<0.05)。
四、讨论FGF21基因作为生长因子家族的一员,在多种生理过程中发挥着重要作用。
本研究利用CRISPR-Cas9技术成功构建了FGF21基因敲除小鼠模型,并发现该基因对毛发生长具有重要作用。
毛发生长受多种基因和环境因素的影响,而FGF21基因的缺失可能导致毛囊发育不良、毛发稀疏、色泽暗淡等现象。
CRISPR/Cas9 是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,可用来对抗入侵的病毒及外源DNA。
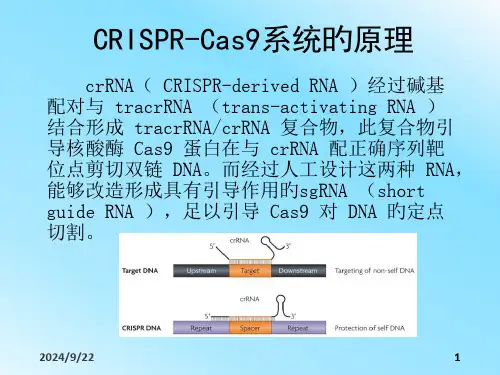
CRISPR/Cas9 系统通过将入侵噬菌体和质粒DNA 的片段整合到CRISPR 中,并利用相应的CRISPR RNAs(crRNAs)来指导同源序列的降解,从而提供免疫性.原理此系统的工作原理是crRNA( CRISPR—derived RNA )通过碱基配对与tracrRNA (trans—activating RNA )结合形成tracrRNA/crRNA 复合物,此复合物引导核酸酶Cas9 蛋白在与crRNA 配对的序列靶位点剪切双链DNA。
而通过人工设计这两种RNA,可以改造形成具有引导作用的sgRNA (singleguide RNA ),足以引导Cas9 对DNA 的定点切割。
作为一种RNA 导向的dsDNA 结合蛋白,Cas9 效应物核酸酶是已知的第一个统一因子(unifying factor),能够共定位RNA、DNA 和蛋白,从而拥有巨大的改造潜力。
将蛋白与无核酸酶的Cas9(Cas9 nuclease-null)融合,并表达适当的sgRNA ,可靶定任何dsDNA 序列,而sgRNA 的末端可连接到目标DNA,不影响Cas9 的结合。
因此,Cas9 能在任何dsDNA 序列处带来任何融合蛋白及RNA,这为生物体的研究和改造带来巨大潜力.应用基因敲除动物模型一直以来是在活体动物上开展基因功能研究、寻找合适药物作用靶标的重要工具.但是传统的基因敲除方法需要通过复杂的打靶载体构建、ES细胞筛选、嵌合体小鼠选育等一系列步骤,不仅流程繁琐、对技术的要求很高,而且费用大,耗时较长,成功率受到多方面因素的限制。
即使对于技术比较成熟的实验室,利用传统技术构建基因敲除大、小鼠一般也需要一年以上。
2013 年1 月份,美国两个实验室在《Science》杂志发表了基于CRISPR-Cas9 技术在细胞系中进行基因敲除的新方法,该技术与以往的技术不同,是利用靶点特异性的RNA 将Cas9 核酸酶带到基因组上的具体靶点,从而对特定基因位点进行切割导致突变。
doi:10.3969/j.issn.1674-5817.2019.01.005利用CRISPR/Cas9技术构建环指蛋白126基因敲除小鼠高梦樵I艾东旭2,李饪打孙菲I,王进I范君文I袁征[刘源I,孙兆增「(1.军事医学研究院实验动物中心,北京100071;2.塔里木大学生命科学学院,阿拉尔市843300)[摘要]目的利用CRISPR/Cas9技术,获得环指蛋白126(RNF126)基因敲除小鼠模型。
方法针对C57BL/6小鼠Rnf\26基因的第四外显子设计敲除引物,构建sgRNA重组表达载体。
通过体外转录获得sgRNA及Cas9mRNA,并以显微注射的方式将RNA导入到C57BL/6小鼠的受精卵中,通过胚胎移植至代孕母鼠。
获得子代工程鼠后,用PCR对其进行鉴定。
结果成功获得针对鼠Rnfl26基因的sgRNA以及Cas9mRNA;通过显微注射获得了82枚状态良好的受精卵并成功移植至3只代孕母鼠体内;获得了11只F0代仔鼠,鉴定后选择一只单链缺失62个碱基的阳性鼠进行扩繁并在Fl、F2代检测到该突变。
结论成功构建Rnf\26基因敲除C57BL/6小鼠,该模型可用于RNF126基因及其表达产物的生物学功能研究。
[关键词]CRISPR/Cas9;环指蛋白126(RNF126);基因敲除;动物模型[中图分类号]Q95-33[文献标志码]A[文章编号]1674-5817(20⑼01-0021-05环指蛋白126(ring finger protein126,RNF126)是一种E3泛素连接酶,与泛素激活酶E1、泛素结合酶E2共同参与真核生物体内特定蛋白的泛素化,从而诱导蛋白的降解。
近期研究g表明,RNF126在许多肿瘤性疾病的发病中起关键性作用。
抑制人类舌癌细胞SCC9与SCC25中RNF126基因的表达可以使癌细胞的增殖能力和活性下降內。
在人乳腺癌组织中RNF126髙度表达,并可同时作为判定侵润性乳腺癌预后不良的生物标记物⑷。
双基因敲除小鼠繁殖工作:CRISPR/Cas9方案构建双基因敲除鼠,得到F0杂合子之后,如何建系才能获得双基因敲除纯合子小鼠?这是经常被问到的问题,下面就简单回答一下。
假设我们的目的基因为A和B,通常用CRISPR/Cas9方法得到的基因敲除鼠为杂合子,双杂合子小鼠基因型为AaBb,大写字母代表野生型(dominant),小写字母代表突变型(recessive)。
得到F0杂合子(AaBb)之后,可以用以下方案之一来获得双基因敲除纯合子小鼠:方案一:1.将双杂合子小鼠(AaBb)与野生鼠(AABB)交配,理论上将得到25%的野生型(AABB),25%基因A单杂合子(AaBB),25%基因B单杂合子(AABb)及25%双杂合子小鼠(AaBb)。
2.将所得到的双杂合子小鼠(AaBb)互交(inter-cross),理论上6.25%的后代将会是双基因敲除纯合子小鼠(aabb),见下图。
3.由于双基因敲除实验中一般都需要单基因敲除动物作为对照,所以在进行上面小鼠breeding的同时可以将基因A单杂合子(AaBB)互交,在后代中鉴定出基因A纯合子(aaBB),同样将基因B单杂合子(AABb)互交,在后代中鉴定出基因B纯合子(AAbb)。
方案二:将双杂合子小鼠(AaBb)与单基因纯合子(如aaBB)交配,所生小鼠中约25%为基因A纯合子而基因B杂合子(aaBb,见下图左)。
然后将aaBb小鼠互交,理论上后代小鼠中25%为双基因敲除纯合子小鼠(aabb),见下图右。
需要特别注意的几个问题:1)上面所讲的方法适用于位于不同的染色体两个基因的基因敲除,如果两个基因位于同一条染色体上,要通过上述方法得到双基因敲除纯合子小鼠很难;2)上述方法有赖于基因特异性的Genotyping PCR assays。
在开始setup breeding之前必须将两个目的基因特异性的Genotyping PCRassays 准备好;3)要事先研究一下目的基因敲除后有无胚胎致死性,是否影响其生长发育等。
《利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系》篇一一、引言基因编辑技术近年来在生命科学领域取得了显著的进展,其中CRISPR-Cas9系统因其高效、精确的基因编辑能力而备受关注。
DUSP9作为一种在细胞信号转导过程中发挥重要作用的基因,对其功能的研究对于揭示生命活动规律、探索疾病发病机制以及新药研发等方面具有重要意义。
本研究旨在利用CRISPR-Cas9系统构建DUSP9基因敲除小鼠胚胎干细胞系,为后续研究提供可靠的实验材料。
二、材料与方法1. 材料(1)小鼠胚胎干细胞(mESCs);(2)CRISPR-Cas9系统相关质粒;(3)DUSP9基因敲除载体;(4)相关实验试剂与仪器。
2. 方法(1)设计并构建DUSP9基因敲除载体;(2)将DUSP9基因敲除载体与CRISPR-Cas9系统相关质粒共转染至mESCs中;(3)筛选并扩增转染后的细胞,获得DUSP9基因敲除的mESCs;(4)对获得的DUSP9基因敲除mESCs进行鉴定与验证。
三、实验结果1. DUSP9基因敲除载体的构建与鉴定通过PCR及测序等方法,成功构建了DUSP9基因敲除载体,并对其进行了鉴定,确认其序列正确无误。
2. CRISPR-Cas9系统介导的DUSP9基因敲除将DUSP9基因敲除载体与CRISPR-Cas9系统相关质粒共转染至mESCs后,通过筛选及扩增,成功获得了DUSP9基因敲除的mESCs。
3. DUSP9基因敲除mESCs的鉴定与验证通过PCR、Western blot及测序等方法,对获得的DUSP9基因敲除mESCs进行了鉴定与验证。
结果表明,DUSP9基因已成功被敲除,且未出现非特异性剪切。
同时,mESCs的生长状况及表型未发生明显改变。
四、讨论本研究利用CRISPR-Cas9系统成功构建了DUSP9基因敲除小鼠胚胎干细胞系。
这一研究成果为研究DUSP9基因的功能提供了可靠的实验材料。
中外医疗China &Foreign Medical Treatment高尿酸血症(hyperuricemia)是由嘌呤代谢紊乱,尿酸排泄障碍引起的血尿酸异常为临床表现的异质性疾病。
尿酸病理性升高有5%~12%的风险导致尿酸结晶累积并损伤关节[1],引起痛风、急性及慢性关节炎等。
并且,高尿酸血症易引发并发症,如高血压、高血脂、2型糖尿病、冠心病。
临床上常规的高尿酸血症及痛风治疗手段为口服降尿酸药物[2],目前尚未有根治痛风的药物,新型降尿酸药物开发需要利用高尿酸动物模型进行降尿酸药物药效及药理筛选。
故建立稳定性好,更接近患者发病特征的高尿酸动物模型,能提高新型降尿酸药物开发效率及成药性,优化痛风治疗格局。
近年来,CRISPR/Cas9基因编辑技术凭借其简便性、特异性和高效性,在疾病动物模型构建领域应用日趋广泛和深入。
该研究利用CRISPR/Cas9,敲除小鼠基因组内尿酸氧化DOI:10.16662/ki.1674-0742.2020.07.032基于CRISPR/Cas9构建小鼠UOX 基因敲除模型张茹君1,夏海林1,朱赟2,黄晶3,孟雨菡21.常州卡文斯实验动物有限公司,江苏常州213104;2.江苏科标医学检测有限公司,江苏常州213161;3.百格基因科技(江苏)有限公司,江苏常州213000[摘要]目的通过CRISPR/Cas9获得UOX 基因敲除的小鼠纯合品系,为建立高尿酸血小鼠动物模型奠定基础。
方法根据小鼠UOX 基因第三外显子前后两个位点设计双sgRNA,通过PCR、体外转录和纯化获得sgRNA 和Cas9mRNA。
将sgRNA 和Cas9mRNA 显微注射进小鼠原核胚后,体外培养至二细胞胚阶段,进行胚胎移植至代孕母鼠。
F0代小鼠出生后,提取其DNA 进行电泳分析和测序分析。
F0代交配繁殖至F2代获得UOX 缺失的小鼠纯合品系。
结果显微注射sgRNA 和Cas mRNA 至原核胚,成功获得50枚囊胚。
使用CRISPR-CAS系统构建可遗传的基因敲除小鼠和大鼠致编辑:CRISPR-CAS系统已经成为一种细胞和模式生物中有效的基因编辑技术。
我们使用CRISPR-CAS系统,通过同时注入两种单导向RNA靶向Uhrf2,同时带入Cas9 mRNA来诱导小鼠的DNA片段缺失。
此外,我们通过一种单一的显微注射方法得到了敲除Mc3R和Mc4R两种基因的大鼠。
在小鼠和大鼠中均观察到较高的种系转移效率(突变可遗传)。
成簇有序间隔短回文重复关联蛋白系统(CRISPR-CAS系统)是一种在细菌和古细菌中演变的针对病毒和质粒入侵的基于RNA的后天免疫系统。
【Bamboo注:该系统由一段Cas基因(双链DNA核酸酶)加一段特异序列组成,Cas作用为结合导向RNA,切割目的基因,导向RNA为CRISPR序列转录而成,有二级结构】根据作用机制不同,CRISPR-CAS系统目前有三种类型。
在第二类型(下文称该系统)中,CRISPR序列转录RNA(crRNA)和反式激活RNA(TraceRNA)结合后有能力引导Cas9核酸内切酶到特定的序列,从而导致目标DNA双链缺失。
【Bamboo注:机理:摄取了病毒DNA后,CRISPR 序列在转录产物tracrRNA,表达产物CAS蛋白,RNA酶共同作用下产生导向RNA,导向RNA含有病毒DNA序列,可遗传给下一代,再遇到病毒DNA时将其剪切】之前的研究表明在哺乳动物中有多种基因工程使用RNA介导的Cas9核酸酶系统。
最近,使用该系统进行高效基因编辑已经在斑马鱼、小鼠和细菌中实现。
几个小组也证明通过该系统介导的在细胞和斑马鱼中基因靶向效率与ZFNs(锌指核酸酶)和TALENs(转录活化因子效应核酸酶)【Bamboo注:另外两种常见DNA编辑方式】相似或较高。
虽然已经有报道在单个小鼠胚胎中可使用该系统打乱多个基因,但是在动物体中尚未见该系统介导的突变种系转移。
此外,长的特异的基因组DNA片段能否被该系统敲除也是未知的。
CRISPR-CAS系统基因打靶在其他哺乳动物模型,例如实验室大鼠的效用,还需要确定。
在这里,俺们报告俺们使用该系统在小鼠和大鼠中实现了高效的,可遗传的基因敲除。
图1使用该系统产生基因突变鼠。
(a)该系统构建:Spacer-核酸引导序列;DR-分隔核酸引导序列的小重复片段;NLS-核酸定位信号序列。
(b) F0代大鼠注射导向RNA后突变体的检测:Mc4r靶向前后(-,+)分别使用T7E1酶切Mc4r F0代大鼠尾基因组PCR产物。
箭头为突变带,M是Marker.(c) Mc3r和Mc4r基因的序列。
红框为核苷酸替换。
右侧显示碱基对序列改变。
通过DNA测序分析六组克隆扩增的PCR产物序列。
各基因型在六组克隆的发生率列在右侧。
为了测试该系统在小鼠中的活动,初始试验选用了细胞中基因组Th位点已经被有效靶向的小鼠。
首先,我们向FVB小鼠品系雄性原核(受精卵核物质融合前)中注入不同浓度的线性DNA(针对特定CrDNA和tracrRNA的人工编码Cas9核酸内切酶基因)(图1a),在子鼠中检测Th位点基因突变情况。
同ZFNs(锌指核酸酶)和TALENs (转录活化因子效应核酸酶)相似,该系统诱导的双链DNA缺失主要被NHEJ(非同源性末端结合修复机制)修复。
通过高浓度(2.5ng/μl)DNA注射,仅有1/11(9%)的子鼠产生Th位点突变,低浓度则只有野生型子鼠(表1和附图1)。
这说明该系统使用线性DNA突变率比较低。
为了提高效率,于是我们注射体外合成的RNA。
首先,我们通过体外转录Cas9片段构建了Cas9表达载体,在包含靶向的人工编码序列载体里隐藏了SP6启动子序列。
我们还建立了一个融合crRNA和tracrRNA的表达载体,使用T7启动子驱动在tracrRNA3‘端衍生合成含有20个靶向特异核苷酸序列的sgRNA(定制合成RNA)(图1a)。
同TALENs同样浓度(25ng/μl)的Cas9 mRNA,还有Th靶向sgRNA(定制合成RNA)(12.5ng/μl)被显微注射到单细胞阶段的C57BL / 6小鼠胚胎的细胞质中。
经过T7E1酶切和DNA测序,90%(8/9)注射后的子鼠Th位点有突变(附图1)。
试验组6最长的缺失有70bp。
在Rheb基因位点插入或缺失突变的试验小鼠也可以通过这个策略高效率得到(表1和附图2)。
CRISPR-CAS系统的一个最重要的优点是,Cas9蛋白可以通过单个gRNAs引导修改多个基因组靶标。
为了在小鼠体内测试这个,我们针对Uhrf2中横跨86bp的两个相邻位点设计并插入了两种sgRNA(定制合成RNA)和Cas9 mRNA到胚胎里,12组F0代小鼠中有11组Uhrf2位点有突变,有6组在同样的等位基因上有7种不同的突变(附图3)。
6组中有3组在两个位点有大量缺失(附图3)。
这些大的突变可能是由于两个位点DNA同时切除,随后又被末端连接。
通过ZFNs和TALENs在试验中所产生的突变可被有效地遗传到下一代,但据我们所知,CRISPR -CAS系统的种系遗传效率在动物中尚未见报道。
为了研究这个问题,我们通过T7E1酶切或DNA测序检测Th试验组和野生型小鼠幼崽或胎儿的基因型。
虽然在DNA注射后鼠尾部DNA的检测中只有两个突变体,但是10组F1代小鼠胎儿中有6组个发现有5种不同的突变(附图4)。
这些数据意味着试验组的突变源自于Cas9在胚胎中切除DNA的延误。
这也表明从试验组6组鼠尾PCR产物的DNA测序中有可能发现不了所有的突变。
通过基因注射产生的另两个试验组突变也被传给下一代(附图4 )。
这些数据表明,CRISPR -CAS系统是有用的遗传工具,产生可遗传的突变小鼠效率很高。
实验室老鼠是模拟疾病的重要工具,是毒理学和药理学的优秀模型。
之前的研究已经通过ZFNs和TALENs成功产生基因敲除小鼠,我们尝试使用该系统产生基因敲除小鼠。
针对大鼠黑素皮质素受体3(MC3R)和黑皮素4受体(MC4R)我们合成了两种sgRNA(定制合成RNA)。
Cas9 mRNA和两种sgRNA的混合物分别被注射进1细胞期的Sprague-Dawley大鼠胚胎,然后将其转移到假孕雌性。
提取子鼠基因组DNA进行PCR 扩增。
T7EI酶切及DNA测序数据表明,无论是MC3R和MC4R基因位点都是由CRISPR -CAS系统修改得到(图1b , c)。
然而,这两个Cas9为基础的核酸酶活性有很大的不同。
通过T7EI酶切及DNA测序数据,15个F0幼鼠中13个幼崽被确定为MC4R基因突变鼠的试验组(图1b)。
不需酶切就很容易用PCR检测含有大量缺失的试验组。
通过T7E1酶切无法鉴定MC3R基因突变试验组,但通过测序得知一只大鼠有MC4R基因突变,也有一个单核苷酸缺失的MC3R基因突变(图1c)。
我们研究了MC4R试验组体重,食物摄取,胰岛素水平和瘦素mRNA水平,发现该突变体与已被用作肥胖动物模型的N -乙基-N-亚硝基脲(ENU )诱导MC4R突变鼠表现出相似的表现型(附图5)。
这些数据表明,CRISPR -CAS系统可以产生基因敲除鼠,并且,该效率取决于靶标基因。
另外,单次注射能够诱导大鼠的至少两个不同基因的混乱。
为了确定Cas9介导的基因突变大鼠种系遗传能力,我们通过MC4R突变鼠试验组12与野生型鼠(杂交?)确定鼠胎儿的MC4R序列。
经鉴定6组胎儿中的3组含有两个不同的突变(附图6),这表明Cas9介导的基因突变大鼠具有高效率的种系遗传。
基因组编辑另一个重要的问题是体内特异性。
以往的研究表明有两个错配如何影响Cas9介导的DNA敲除的规则。
其一在PAM的5‘端的单碱基到12个bp的错配,完全取消了Cas9介导的DNA缺失。
另一种是如果要高效清除,gRNA和PAM近端的靶标向DNA之间需要至少13个bp的匹配,并且该序列以外的不匹配是允许的。
我们通过分析这两类小鼠基因组中潜在的脱靶位点研究了该系统对靶标的特异性。
因为分析所有潜在的脱靶位点比较难,我们选定少于四个错配的两个部位或与相邻的PAM匹配大于11个bp(附图7)的,通过测序观察到在12个试验组的潜在脱靶位点没有突变(附图7)。
在细胞和斑马鱼中, CRISPR - CAS系统介导的基因混乱同TALENs有类似的效果。
但是我们的研究,同之前的研究一致表明该系统比ZFNs和TALENs更牛掰,至少在小鼠中。
我们之前的研究发现TALENs产生的小鼠效率为13%到67%,而该系统诱导效率通常大于70%。
我们还注意到,CRISPR-CAS系统(表1)的毒性(指注射后立即生存能力)比TALENs有一点点大。
CRISPR-CAS系统的种系遗传效率与TALENs相似。
我们也发现与TALENs相比,某些靶标不能由CRISPR-CAS敲除,原因不明(未示出数据),但是潜在的靶标应该比TALENS更多。
虽然靶标比TALENS和ZFNs的短,但我们的研究中使用CRISPR-CAS系统没有脱靶突变事件表明它是一个可靠的动物基因编辑技术。
Fu最近出版的一篇文章也更全面的研究了各基因编辑系统的优劣。
在这项研究中,我们使用CRISPR-CAS系统成功地产生了不同遗传背景特异基因敲除小鼠,在Sprague-Dawley大鼠中进行了特异基因敲除。
在哺乳动物的有机体功能基因研究方面,高效率的基因组修饰活性和种系遗传效率的RNA引导的CRISPR-CAS 系统是很有用的基因工具。