16实验十六 复方磺胺甲恶唑片的含量测定
- 格式:doc
- 大小:68.00 KB
- 文档页数:3


双波长分光光度法测定复方磺胺甲噁唑片中两组分的含量王乐婷;郭秀琴;李珂【摘要】Objective:A dual wavelength determination of sulfamethoxazole and trimethoprim in compound sulfamethox-azole tablets by spectrophotometry. Methods:Determined by dual wavelength spectrophotometry. Wavelength determination of sulfamethoxazole was 257nm,the reference wavelength was 305nm. Taking 0. 4% sodium hydroxide solution as SMZ blank solvent. Wavelength determination of trimethoprim was 239nm,the reference wavelength was 293 nm. Hydrochloric acid potassium chloride solution as the TMP blank solvent. Results:The results showed that both the standard curve of sul-famethoxazole concentration ranging from 2 to 10 μg·ml - 1 and trimethoprim concentration ranging from 0. 2 to 1. 0 μg· ml - 1 was in correspondence with Beer′s law. The absorptivity difference and the concentration showed a good linear relation-ship. The average recoveries and RSD were 97. 84% 、0. 25% for sul-famethoxazole,and were 97. 91% 、0. 2% for trime-thoprim. Conclusion:The method is simple and accurate,which can be used as one of methods for quanlity control of the tables.%目的:建立复方磺胺甲噁唑片中磺胺甲噁唑和甲氧苄啶含量测定的双波长分光光度法。


验证性试验实验十八复方磺胺甲噁唑片中磺胺甲恶唑的含量测定一、实验目的1.掌握双波长法的基本原理。
2.掌握复方制剂不经分离直接测定各组分含量的方法。
3.熟悉紫外分光光度仪的使用。
二、仪器与试药1.仪器Mettler AL204电子天平 752型紫外-可见分光光度仪研钵定量滤纸(直径10cm)胖肚移液管规格:25mL容量瓶规格:25mL 、100mL 刻度移液管规格:10mL2.试药复方磺胺甲噁唑片规格:含磺胺甲噁唑(SMZ) 0.4g、甲氧苄啶(TMP) 0.08g95%乙醇氢氧化钠硫酸三、实验原理1.双波长分光光度法(1)双波长分光光度法消除干扰吸收的基本原理在干扰组分的吸收光谱上吸收系数相同的两个波长处,若被测组分的吸收系数有显著差异,则可用于消除干扰吸收,即直接测定混合物在此两波长处的吸收度之差值,该差值与被测物浓度成正比,而与干扰物浓度无关。
用数学式表达如下:ΔA(λ1λ2)=A a+b1-A a+b2=A a1-A a2+A b1-A b2=ΔE.a C a.L+ΔE b.C b.L若b为干扰物,所选波长λ1λ2处的E b相等,所以ΔE b=0则ΔA混=ΔE a.C a.L与干扰物浓度无关。
(2)双波长法测定复方磺胺甲噁唑片中SMZ含量的原理复方磺胺甲噁唑片是含磺胺甲基异噁唑(SMZ)及甲氧苄啶(TMP)的复方制剂。
在0.1mol/L 氢氧化钠液中,SMZ在257nm波长处有一最大吸收峰,TMP在257nm和304nm波长处为等吸收点,而SMZ在这两波长处的吸收度差异大,见下图。
所以测得样品在257nm和304nm波长处的吸收度差值ΔA与SMZ浓度成正比,与TMP浓度无关。
四、实验内容1.工作曲线的制备(1)标准贮备液的配制分别精密称取磺胺甲基异噁唑对照品0.1g ,置100mL 容量瓶中,加入50m L 95%乙醇溶解后,以0.1mol/L 氢氧化钠液稀释至刻度,吸取10mL 置100mL 容量瓶中,以0.1mol/L 氢氧化钠液稀释至刻度,得标准贮备液(100μg /mL )。

续表1序号保留时间t /min化合物名称分子式分子量相对含量(%)79.3213-octadecenoic acid(Z)(Z)-13-十八碳烯酸C 18H 34O 228221.6289.73octadecan oic acid 十八烷酸C 18H 36O 228433.46910.6210,13-octadecadienoic acid 10,13-十八碳二烯酸C 18H 32O 22800.131010.7910-noadecen oic acid 10-十九烯酸C 19H 36O 22960.091111.26noadecanoic caid 十九烷酸C 19H 38O 22980.281212.7211-eicosenoic acid 11-二十碳烯酸C 20H 38O 2310 2.101313.23eicosanoic acid 二十碳烷酸C 20H 40O 2312 1.771417.42docosanoic acid 二十二碳烷酸C 22H 44O 23400.18由表1可以看出,从中药材木鳖子中共鉴定出14种脂肪酸,占脂肪酸总含量的89.23%,其中饱和脂肪酸7种,占总脂肪酸含量的47.32%不饱和脂肪酸7种,占脂肪酸总量的41.91%。
3 讨论木鳖子中不饱和脂肪酸以亚油酸(19.85%)、(Z)-13-十八(碳)烯酸(21.62%)、11-二十(碳)烯酸(2.10%)为主。
近年来的研究表明不饱和脂肪酸对人体有降低血脂、胆固醇和血压,抗血栓、抗动脉硬化,预防心血管疾病,增强记忆力,预防老年痴呆症,防癌等多种作用[3]。
木鳖子是一种常见的中药,对中药木鳖子的研究已较深入,但对其脂肪酸的研究还未见报道。
本实验方法具有简单、准确、脂肪酸检出率高等优点,其结果将对木鳖子的深层次的开发和应用提供科学依据。
参考文献:[1] 宋立人,洪 恂,丁绪亮,等.现代中药大辞典,上册[M ].北京:人民出版社:349-351.[2] S.R..Heller ,G.W.A.M iline.EPA/NIH M ass Spectral date[M ].U .S.Washington D.C.,1978:1-4.[3] 周永红.火麻仁油中脂肪酸的GC-M S 分析[J ].中国油脂,2004,29(3):72-72.收稿日期:2004-11-12; 修订日期:2005-02-12作者简介:熊久林(1956-),男(汉族),湖北黄梅人,现任湖北省黄石市药品检验所副主任药师,主要从事药品检验工作.高效液相色谱法测定复方磺胺甲唑片中磺胺甲唑和甲氧苄啶的含量熊久林,孙仲葆,黎 源,马锦星,运 委,张 晶(湖北省黄石市药品检验所 435000)摘要:目的:建立同时测定复方磺胺甲唑片中磺胺甲唑和甲氧苄啶含量的高效液相色谱法。

![[重点]复方磺胺甲恶唑的含量测定](https://uimg.taocdn.com/4b4157c13086bceb19e8b8f67c1cfad6195fe9cd.webp)
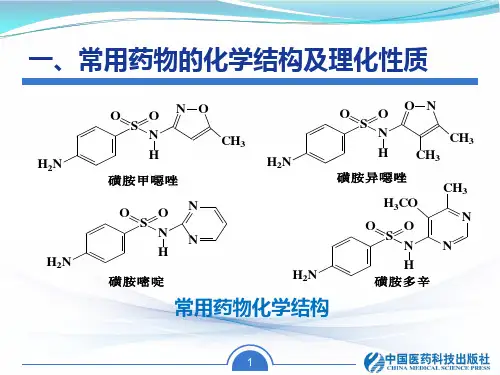
高效液相色谱法测定复方磺胺甲噁唑片的含量[实验目的]1掌握高效液相色谱法测定复方磺胺甲噁唑片的含量的基本原理2掌握高效液相色谱仪的操作及注意事项[实验原理]结构特点:苯环(紫外吸收)、芳伯胺基(重氮化-偶合反应)、磺酰胺基上的活泼氢有一定酸性,可与碱成盐或与重金属离子反应。
分子式:C10H11N3O3S 分子质量:253.28。
本品为白色结晶性粉末。
熔点167℃,易溶于稀盐酸;氢氧化钠溶液或氨水,几乎不溶于水。
无臭,味微苦。
外标法是以待测组分的纯品作为对照品,以供试品中待测组分的峰面积或峰高进行定量分析,本实验采用峰面积进行定量。
分别精密量取一定量的对照品和供试品配制成溶液,分别进相同的体积的对照品溶液和供试品溶液,在完全相同的色谱条件下进行色谱分析,测定峰面积。
[实验仪器、试剂]仪器:75-4型紫外可见分光光度计 (上海分析仪器厂 ),高效液相色谱仪,色谱柱,超声波清洗仪,紫外吸收检测器,无油隔膜真空泵,抽滤装置,电子分析天枰,称量纸,研钵,容量瓶(100ml,50ml,25ml,10ml),漏斗,滤纸,烧杯,玻璃棒,移液枪试剂:复方磺胺甲噁唑片(山东新华制药股份有限公司 ,批号 0412073 ),甲氧苄啶,磺胺甲噁唑,甲醇(色谱纯),水(超纯水),乙腈(色谱纯),三乙胺 (色谱纯),醋酸溶液(1→100),[实验步骤]1.色谱条件与系统适用性试验:填充剂:十八烷基硅烷键合硅胶为填充剂流动相:水-乙腈-三乙胺(799∶200∶1)(用醋酸溶液(1→100)调节pH值至5.9±0.1)检测波长:254nm流速:1.0mL/min进样量:20ul理论塔板数:按甲氧苄啶计算应不低于2000,甲氧苄啶与磺胺甲噁唑峰之间的分离度应大于5.0。
甲氧苄啶峰与磺胺甲噁唑峰的拖尾因子均不得过2.0。
2.测定法2-1溶液的配制2-1-1对照品储备液的配制:取磺胺甲噁唑200mg,精密称定,置50ml容量瓶中,加甲醇溶解并稀释至刻度,摇匀,备用。
目的:为检验复方磺胺甲噁唑片中间产品规定一个标准的程序,以便获得准确的实验数据。
范围:适用于复方磺胺甲噁唑片中间产品的检验。
职责:检验员,检验室主任对本规程实施负责。
规程:1 性状:本品为白色片.2 鉴别:2.1 试剂与仪器2.1.1 稀盐酸 2.1.2 0.1mol/L亚硝酸钠溶液2.1.3 碱性B-萘酚试液 2.1.4 稀硫酸2.1.5 甲醇 2.1.6 氯仿-甲醇-二甲基酰胺(20:2:1)2.1.7 碘试液 2.1.8 硅胶GF254薄层板2.1.9 烧杯、量筒、试管 2.1.10 微量进样器2.1.11 层析缸 2.1.12 漏斗、漏斗架2.1.13 电炉2.2 项目与步骤2.2.1 取本品细粉适量(约相当于磺胺甲噁唑50mg),显芳香第一胺的鉴别反应:取供试品约50mg,加稀释盐酸1ml,必要时缓缓煮沸便溶解,放冷,加0.1mol/L亚硝酸钠溶液数滴,滴加碱性B-萘酚试液数滴,视供试品不同,生成由橙黄到桔红色,为符合规定。
2.2.2 取本品的细粉适量(约相当于甲氧苄啶50mg),加稀硫酸10ml微热溶解后,放冷、滤过、滤液加碘试液0.5ml,即生成棕褐色沉淀,为符合规定。
2.2.3 取本品的细粉适量(约相当于磺胺甲噁唑0.2g),加甲醇10ml,振摇,滤过,取滤液作为供试品溶液,另取磺胺甲噁唑0.29g、甲氧苄啶40mg,加甲醇10ml溶解,作为对照溶液;照薄层色谱法 (SOP-QC-304-00)试验,吸取上述两种溶液各5ml,分别点于同一硅胶GF254薄层板上,以氯仿-甲醇-二甲基甲酰胺(20:2:1)为展开剂,展开后,晾干,置紫外光灯(254nm)检视,供试品溶液所显两种成分的主斑点的位置应与对照溶液的主斑点相同为符合规定。
3 检查:3.1 试剂与仪器3.1.1 电子天平(万分之一克) 3.1.2 片剂崩解仪3.1.3 片剂脆碎度仪 3.1.4 吹风机3.2 项目步骤3.2.1 崩解时限:取本品6片,照崩解时限检查法(SOP-QC-330-00) 检验,应在15分钟内全部崩解,如有1片崩解不完全,应另取6片用样方法复试,全部崩解完全为符合规定。

XXXXXXXXX有限公司复方磺胺甲噁唑片中间产品(素片)检验操作规程1. 性状取本品,外观检查应为白色片、完整光洁、色泽均匀。
2. 检查2.1 重量差异2.1.1 仪器与用具分析天平(感量0. 1mg),称量瓶。
2.1.2 操作步骤2.1.2.1 照重量差异检查法项下方法,取本品20片,精密称定总重量,求得平均片重。
2.1.2.2 再分别精密称定各片的重量,与平均片重相比较。
2.1.3 计算2.1.3.1 平均片重 = 20片总重量/20。
2.1.3.2 允许片重差异范围 = 平均片重±平均片重×重量差异限度(±5%)。
2.1.4 结果与判定本品的重量差异限度为±5%。
超出重量差异范围的药片不得多于2片,并不得有1片超出限度1倍。
2.2 崩解时限2.2.1 仪器与用具升降式崩解仪,1000ml烧杯,温度计(分度值1℃)。
2.2.2 操作步骤2.2.2.1 照片剂崩解时限检查法项下方法,将升降式崩解仪的吊篮通过上端的不锈钢轴悬挂于金属支架上,浸入1000ml烧杯中,并调节吊篮位置使其下降时筛网距烧杯底部25mm,烧杯内盛有温度为37℃±1℃的水,调节水位高度使吊篮上升时筛网在液面下15mm处;2.2.2.2 取本品6片,分别置吊篮的玻璃管中,启动崩解仪进行检查。
2.2.3 结果与判定2.2.3.1各片均应在15分钟内全部崩解;2.2.3.2如有1片崩解不完全,应另取6片,按上法复试,均应全部崩解。
2.2.4 注意事项2.2.4.1 在测试过程中,烧杯内的水温应保持 37℃±1℃。
2.2.4.2 每测试一次后,应清洗吊篮的玻璃管内壁及筛网、档板等,并重新更换水。
2.3 脆碎度2.3.1仪器与用具脆碎度检查仪,分析天平(感量0.1 mg),吹风机,称量瓶。
2.3.2 操作步骤2.3.2.1 照片剂脆碎度检查法项下方法,调节脆碎度检查仪的转数每分钟25±1转,设定试验时间为4分钟,则片剂滚动的总数次为100次;2.3.2.2 取本品若干片(约相当于总重量6.5g),用吹风机吹去脱落的粉末;2.3.2.3 取空称量瓶,精密称定,将上述本品置称量瓶中,精密称定,两次称量之差即为本品的重量;2.3.2.4 将上述称定重量后的本品置脆碎度检查仪圆筒中,开动仪器电动机进行检查试验;2.3.2.5 取试验结束后的本品外观检查,不得出现断裂、龟裂或粉碎现象;2.3.2.6 外观检查完毕后,再用吹风机吹去粉末后,置上述称定重量的称量瓶中,精密称定,两次称量之差值即为试验后本品的重量。

双波长法测定复方磺胺甲噁唑含量之迟辟智美创作一、目的1.掌握复方制剂的分析特点及赋形剂的干扰与排除方法.2.掌握双波长分光光度法测定复方磺胺甲噁唑片中磺胺甲噁唑与甲氧苄啶含量的原理与方法.二、实验内容取本品20片,精密称定,研细,精密称取适量(约相当于磺胺甲噁唑50mg(20片平均片重的8分之一)与甲氧苄啶10mg(20片平均片重的8分之一)),置于100ml量瓶中,加乙醇适量,振摇15分钟磺胺甲口恶唑与甲氧苄啶溶解,加乙醇稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液.精密称取105℃干燥至恒重的磺胺甲噁唑对比品50mg(按原料药95%含量计)与甲氧苄啶对比品10mg(按原料药95%含量计),分置100ml量瓶中,各加乙醇溶解并稀释至刻度,摇匀,分别作为对比品溶液(1)与对比品溶液(2).3.磺胺甲噁唑的测定精密量取供试品溶液与对比品溶液(1)、(2)各2ml,分置100ml量瓶中,各加0.4%氢氧化钠溶液稀释至刻度,摇匀.取对比品溶液(2)的稀释液;以257nm为测定波长(λ2),在304nm波长附近(每间隔0.5nm)选择等吸收点波长为介入波长(λ1),要求△A=Aλ2-Aλ1=0.再在λ2与λ1波长处罚别测定供试品溶液的稀释液与对比品溶液(1)的稀释液的吸收度,求出各自的吸收度差值(△A),计算,即得.精密量取上述供试品溶液与对比品溶液(1)(2)各5ml,分置100ml量瓶中,各加盐酸-氯化钾溶液[取盐酸液(0.1mol/L)75ml与氯化钾,加水至1000ml摇匀] 稀释至刻度,摇匀.取对比品溶液(1)的稀释液,以239.0nm为测定波长(λ2),在295nm波长附近(每间隔0.2nm)选择等吸收点波长为参比波长(λ1),要求△A=Aλ2-Aλ1=0.再在λ2与λ1波长处罚别测定供试品溶液的稀释液与对比品溶液(2)的稀释液的吸收度,求出各自的吸收度差值(△A),计算,即得.本品每片中含磺胺甲噁唑(C10H11N3O2S)应为0.360~0.440g,含甲氧苄啶(C14H12N4O3)应为72.0~88.0mg.三说明1.磺胺甲噁唑与甲氧苄啶的结构分别为:SMZ与TMP的紫外吸收图谱分别为:1 TMP(2.0μg/ml);2 SMZ(10.0μg/ml) 1 TMP(5.0μg/ml);2 SMZ(25.0μg/ml)3 SMZ+TMP;4 辅料3 SMZ+TMP; 4 辅料2.复方磺胺甲噁唑片的处方为:磺胺甲噁唑400g甲氧苄啶80g制成1000片3.本实验采纳双波长分光光度法测定SMZ和TMP的含量.由于干扰组分在测定波长和参比波长处吸收度相等,可在计算△A时消去,所以应用本法可以不经分离,直接测定复方制剂中SMZ和TMP的含量.四、思考题1.试简述双波长分光光度法的原理.2.试查阅本品的其它分析方法,从中了解复方制剂分析的特点及发展趋势.注意:1、取20片后要算一片的平均重量,然后根据这一片来取八分之一重量.2、1,2步伐作为母液配制,可以叫专门三组完成,3步伐由双数组完成,4步伐由双数组完成.3、利用紫外测按时每次换一个波长就要将空白对比液吸收值重新归零,防止呈现负数值情况.。
复方磺胺甲噁唑片汉语拼音Fufang Huang an Jia ezuo Pian英文名Compound Sulfamethoxazole Tablets药物组成磺胺甲噁唑400g,甲氧苄啶80g,/制成1000片,性状本品为白色片。
鉴别(1)取本品的细粉适量(约相当于甲氧苄啶50mg),加稀硫酸10ml,微热溶解后,放冷,滤过,滤液加碘试液0.5ml,即生成棕褐色沉淀。
(2)取本品的细粉适量(约相当于磺胺甲噁唑0.2g),加甲醇10ml,振摇,滤过,取滤液作为供试品溶液;另取磺胺甲噁唑0.2g与甲氧苄啶40mg,加甲醇10ml溶解,作为对照溶液。
照薄层色谱法(附录ⅤH)试验,吸取上述两种溶液各5μl,分别点于同一硅胶GF254薄层板上,以三氯甲烷-甲醇-二甲基甲酸胺(20:2:1)为展开剂,展开,晾干,置紫外光灯(254nm)下检视。
供试品溶液所显两种成分的主斑点的颜色与位置应与对照溶液的主斑点相同。
(3)在含量测定项下记录的色谱图中,供试品溶液两主峰的保留时间应与对照品溶液相应的两主峰的保留时间一致。
(4)取本品的细粉适量(约相当于磺胺甲噁唑50mg),显芳香第一胺类的鉴别反应(附录Ⅲ)。
以上(2)、(3)两项可选做一项。
备注:(1)为甲氧苄啶的鉴别,(4)为磺胺甲噁唑的鉴别,(2)和(3)为甲氧苄啶与磺胺甲噁唑的综合鉴别,详见收藏。
检查溶出度取本品,照溶出度测定法(附录ⅩC第二法),以0.1mol/L盐酸溶液900ml为溶出介质,转速为每分钟75转,依法操作,经30分钟时,取溶液适量,滤过,精密量取续滤液10μl,照含量测定项下的方法,依法测定,计算每片中磺胺甲噁唑和甲氧苄啶的溶出量。
限度均为标示量的70%,应符合规定。
其他应符合片剂项下有关的各项规定(附录ⅠA)。
含量测定照高效液相色谱法(附录ⅤD)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以水-乙腈-三乙胺(799:200:1)(用氢氧化钠试液或冰醋酸调节pH值至5.9)为流动相;检测波长为240nm。
暴露资料,故难以与环境致病因子进行综合分析.本研究对我国食管癌高发的河南省林州市食管癌标本中p 53基因第五外显子进行了DNA 序列测定,突变发生率为20%,突变大多发生在G ∶C 碱基对,G ∶C ※A ∶T 的置换最多,占5/9,A ∶T ※T ∶A 的颠换占3/9,G ∶C ※C ∶G 的占1/9.从突变类型看,10个突变中9个为点突变,1个为插入突变,与文献报道基本一致.突变位点较分散,可能与该地区多种危险因素暴露有关.这一结果与我室前期研究相吻合[4].既往宏观流行病学研究发现,食管癌家族史、食用酸菜不仅是林州市食管癌的危险因素,也与p 53蛋白高表达密切相关[5].我们的研究结果显示,p 53基因第五外显子突变与性别、年龄无关,与食管癌家族史、吸烟也未见关联,可能与本研究分组后样本量尚少有关.但与食用酸菜有明显联系,其OR 值为5.95(1.13~31.26).酸菜中含有包括亚硝酸胺在内的多种致癌的亚硝基化合物,p 53基因突变可能由于包括酸菜在内的多种外源性致癌物的暴露而诱发,从而诱导肿瘤形成.由于DNA 测序费用较高,在经费有限的情况下,本研究仅对p 53基因第五外显子进行了检测和分析,尚不能全面反映p 53基因的突变情况及影响因素,但为进一步研究奠定了基础.【参考文献】[1]Bennett WP ,Hol lstein M C ,M etcalf R A ,W elsh JA ,He A ,ZhuSM ,Kusters I ,Resau JH ,T rump BF ,Lane DP .p 53mutation and protein accumulation during multistage human esophageal Carcino -genesis [J ].Canc er Res ,1992;52(21):6092-6097.[2]Hussain SP ,Harris CC .p 53mutation spectrum and load :The gen -eration of hypotheses linking the exposure of endogenous or exoge -nous carcinogens to human cancer [J ].Mutation Res ,1999;428:23-32.[3]Zheng ZY ,Wang LD ,Shi Stephanie T ,Yang GY ,Xue ZH ,GaoS ,Li YX ,Yang CS .p 53gene mutation in multifocal esophageal precancerous and cancerous les ions in patiens w ith esophageal cancer in highrisk Northern China [J ].Sh ijie Huaren Xiaohua Zaz hi (Wor ld Chin J Digestol ),1999;7(4):280-284.[4]Li LS ,Sun CS ,Zhang XL ,Qiao GB ,Xu DZ ,Han CL ,YangWX ,Chang GS ,Yan M X ,Wang Y ,Zhang H Y .A comparative molecular epidemiological study on esophageal cancer between Xi an and Linzhou [J ].Jiefangjun Yu fang Y ixue Zazhi (Prevet Med PLA ),1999;17(4):255-258.[5]Zhang XL ,Li LS ,Xu DZ ,Sun CS ,Yang WX ,Zhao HZ ,ChangGS ,Yan M X ,Wang Y .A cas e -control study on relationship be -tw een overexpression of p 53protein and esophageal cancer in Linxi -an [J ].Di -si Junyi Da xue X uebao (J Fourth Mil Med Univ ),1997;18(4):330-332.编辑 何扬举收稿日期:2003-04-17; 修回日期:2003-05-01作者简介:何树芸(1952-),女(汉族),陕西省西安市人.副主任药师.Tel .(029)5239845·经验交流· 文章编号:1000-2790(2003)14-1330-01荧光光度法测定复方磺胺甲恶唑片中的磺胺甲恶唑含量何树芸1,冯 云2,郭欢迎1(1陕西省药品检验所,陕西西安710061,2西安市药品检验所,陕西西安710054)【关键词】荧光光度法;复方磺胺甲恶唑片;磺胺甲恶唑【中图号】R927.2 【文献标识码】B 0 引言 复方磺胺甲恶唑片为《中国药典2000年版二部》所载的品种.临床运用较为广泛.其中磺胺甲恶唑的含量测定,药典采用双波长分光光度法,操作费事且要求条件较高.我们利用荧光光度法测定磺胺甲恶唑含量结果令人满意.1 一般资料1.1 仪器和试药 RF -540荧光分光光度计(日本岛津).磺胺甲恶唑对照品由中国药品生物制品鉴定所提供(批号98-2);复方磺胺甲恶唑片样品由陕西省西安妇幼制药厂提供;其他试剂均为分析纯.1.2 对照溶液的配制 取磺胺甲恶唑对照品12.5mg ,精密称定,置100mL 容量瓶中,加0.2mol ·L -1硫酸甲醇液溶解并稀释至刻度.摇匀取出适量,用0.2mol ·L -1硫酸甲醇液稀释成0.5μg ·mL -1即可.1.3 样品溶液的配制 取样品适量,精密称定(相当于含磺胺甲恶唑30mg ),置200m L 量瓶中,加0.2mol ·L -1硫酸甲醇液溶解并稀释至刻度.摇匀、过滤.弃去初滤液,取续滤液适量,用0.2mo l ·L -1硫酸甲醇液稀释成与对照溶液一致的浓度.1.4 标准曲线的制备 精密吸取对照溶液,0,0.2,0.4,0.6,0.8和1.0m L 分别置10m L 量瓶中各加1mol ·L -1的硫酸液和10mL ·L -1邻苯二甲醛液各2mL ,摇匀,在60℃水浴中加热30min .放冷后用甲醇稀释至刻度.摇匀,测定其激发光谱和发射光谱.结果激发波长为365nm ,发射波长425nm .在此条件下,测定其荧光光谱.以对照溶液的浓度为横坐标,荧光强度为纵坐标作回归曲线.回归方程为:Y =16.1+13.8X ,r =0.997(n =3),可见磺胺甲恶唑浓度在0.01~0.05μg ·mL -1范围内浓度与荧光强度的线性关系良好.1.5 精密度实验 精密吸取对照溶液1mL 共4份分别置10mL 量瓶中,照2.3法测定,按测定的荧光强度计算其精密度RSD =1.3%(n =4).其结果表明该方法精密度良好.1.6 回收试验 按处方配制成复方磺胺甲恶唑片的粉末,精密称取一定量,照2.3法测定,计算其回收率.平均回收率为98.5%(n =4),RSD =1.0%.样品其他成分与辅料均无干扰.1.7 供试品的测定 精密吸取样品溶液1mL ,置10mL 量瓶中,照2.3法测定,计算样品的含量(表1).表1 供试品测定结果(n =3)批号荧光法(g /片)药典方法(g /片)20020405010.3760.38120020605020.3860.38920020104010.3790.3902 讨论 用荧光法测定复方磺胺甲恶唑片中磺胺甲恶唑含量与双波长分光光度法测定结果基本一致,说明此方法准确可行.荧光法的灵敏度高,因而对试剂要求也高,故所用的甲醇需加一定量的金属钠,放置4h 后经重蒸馏即可.样品稀释后,在1h 内测定其荧光强度.否则,结果偏低.编辑 黄良田1330第四军医大学学报(J F our th M il M ed Univ )2003;24(14)。
实验九复方磺胺甲噁唑片的含量测定
实验目的:
1、掌握双波长分光光度法消除干扰的原理和波长选择原则。
2、掌握紫外-标准对照法测定药物含量及计算方法。
3、熟悉紫外分光光度仪的构造和使用操作。
实验原理:
复方磺胺嘧啶片系由磺胺嘧啶和甲氧苄啶组成的复方制剂。
两者在紫外区有较强的吸收。
在盐酸溶液(9→1000)中,磺胺嘧啶在308nm处有吸收,而甲氧苄啶在此波长处无吸收,故可在此波长处直接测定磺胺嘧啶的吸收度而求得含量。
甲氧苄啶在277.4nm波长处有较大吸收,而磺胺嘧啶在277.4nm处与308nm处有等吸收点。
故可采用双波长法以277.4nm 为测定波长,308nm为参比波长,测定甲氧苄啶在该两波长处的ΔA(ΔA=A277nm-A308nm)值来计算含量。
复方氨基比林注射液又称安痛定注射液,系氨基比林、安替比林和巴比妥组成的复方制剂,采用双波长分光光度法测定安替比林的含量。
根据巴比妥在酸性溶液中不呈解离状态,而无明显紫外吸收;安替比林在233nm处有较强的吸收;氨基比林在233nm和268nm处有等吸收,选择安替比林的吸收峰波长233nm为测定波长λ1,氨基比林的等吸收波长268nm为参比波长λ2,则△A =A233nm-A268nm只与安替比林的浓度有关,而巴比妥、氨基比林及其他辅料不干扰测定。
实验器材:
试药:磺胺嘧啶对照品,甲氧苄啶对照品,复方磺胺嘧啶片,安替比林对照品,安痛定注射液。
仪器:紫外分光光度仪,石英比色皿,100mL容量瓶,移液管。
实验内容与方法:
(一)复方磺胺嘧啶片(Compound Sulfadiazine Tablets)
本品每片中含磺胺嘧啶(C10H10N4O2S)应为0.360~0.440g;含甲氧苄啶(C14H18N4O3)应为45.0~55.0mg。
[处方]
[含量测定]
磺胺嘧啶取本品10片,精密称定,研细,精密称取适量(约相当于磺胺嘧啶0.2g),置100mL量瓶中,加0.4%氢氧化钠溶液适量,振摇使磺胺嘧啶溶解,并稀释至刻度,摇匀,滤过,精密量取续滤液2mL,置另一100mL量瓶中,加盐酸溶液(9→1000)稀释至刻度,摇匀,照分光光度法(附录),在308nm的波长处测定吸收度;另取105℃干燥至恒重的磺胺嘧啶对照品适量,精密称定,加盐酸溶液(9→1000)溶解并定量稀释制成每1mL中约含40μg的溶液,同法测定;计算,即得。
甲氧苄啶精密称取上述研细的细粉适量(约相当于甲氧苄啶40mg),置100mL量瓶中,加冰醋酸30mL振摇使甲氧苄啶溶解,加水稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;另精密称取甲氧苄啶对照品40mg与磺胺嘧啶对照品约0.3g,分置100mL量瓶中,各加冰醋酸30mL溶解,加水稀释至刻度,摇匀,前者作为对照品溶液(1),后者滤过,取续滤液作为对照品溶液(2)。
精密量取供试品溶液与对照品溶液(1)、(2)各5mL,分置100mL 量瓶中,各加盐酸溶液(9→1000)稀释至刻度,摇匀,照分光光度法测定。
取对照品溶液(2)的稀释液,以308.0nm为参比波长λ1,在277.4nm波长附近(每间隔0.2nm)选择等吸收点波长为测定波长(λ2),要求ΔA=Aλ2-Aλ1=0。
再在λ2和λ1波长处分别测定供试品溶液的稀释液与对照品溶液(1)的稀释液的吸收度,求出各自的吸收度差值(ΔA),计算,即得。
(二)、复方氨基比林注射液(Compound Aminopyrine Injection)
本品每1mL注射液中含氨基比林(C13H17N3O)应为47.5~52.5mg,含安替比林(C11H12N2O)应为18.0~22.0mg,含巴比妥(C8H12N2O3)应为8.55~9.45mg。
[处方]
[含量测定]精密量取本品注射液1mL,用0.1mol/L盐酸稀释至100mL,摇匀,精密量取此液2mL,用0.1mol/L 盐酸稀释至100mL,摇匀,即得供试液。
另精密称取安替比林对照品约0.08g,用0.1mol/L 盐酸溶解并稀释至100mL,摇匀,精密量取此液1mL,用0.1mol/L 盐酸稀释至100mL。
摇匀,即得对照液(约为8μg/mL)。
实验注意事项:
1. 石英比色皿的正确使用和吸收度校正。
2. 吸收度读数三次,取平均值计算含量。
3. 读数后及时关闭光闸以保护光电管。
4. 片剂取样量应是根据平均片重和片剂规格量,计算出来的相当于规定量主药的片
粉重量(片粉重量=平均片重 / 标示量×规定的取样量)。
5. 片剂的含量计算(相当于标示量的百分含量)。
根据药品获得的难易情况,任选一个内容进行实验。