
1.测定波长的选择
对照品溶液的制备:取盐酸莫西沙星对照品适量,精密称定,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,摇匀,滤过,取续滤液作为对照品溶液;
空白辅料溶液的制备:称取按处方比例废纸的空白辅料适量,同对照品溶液方法制备,即得空白辅料溶液。
以盐酸溶液(0.9→1000)为空白,取上述配制的两种溶液照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在200nm~400nm波长范围内扫描测定,记录紫外吸收图谱。
由紫外吸收可知盐酸莫西沙星在295nm左右波长处由最大吸收。空白辅料在此波长处无吸收,不干扰测定。
线性关系考察
精密称取盐酸莫西沙星对照品约22mg,置100ml量瓶中,加盐酸溶液(0.9→1000)溶解并稀释至刻度,摇匀,作为母液,取母液0.5ml、0.8ml、1.0ml、1.2ml、1.5ml分别置50ml量瓶中,加盐酸溶液(0.9→1000)制成每1 ml中约含2μg、3μg、4μg、5μg、6μg的溶液;照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在293nm的波长处测定吸收度,结果见表。以盐酸莫西沙星浓度为横坐标,吸收度为纵坐标,绘制标准曲线,结果在2.2μg/ml~
6.6μg/ml范围内线性良好。回归方程:y = 0.1052x + 0.0142;相关系数r= 0.9999。
精密度试验
精密称取盐酸莫西沙星对照品适量,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在293nm的波长处测定吸收度,重复测定5次,计算RSD。结果见表。
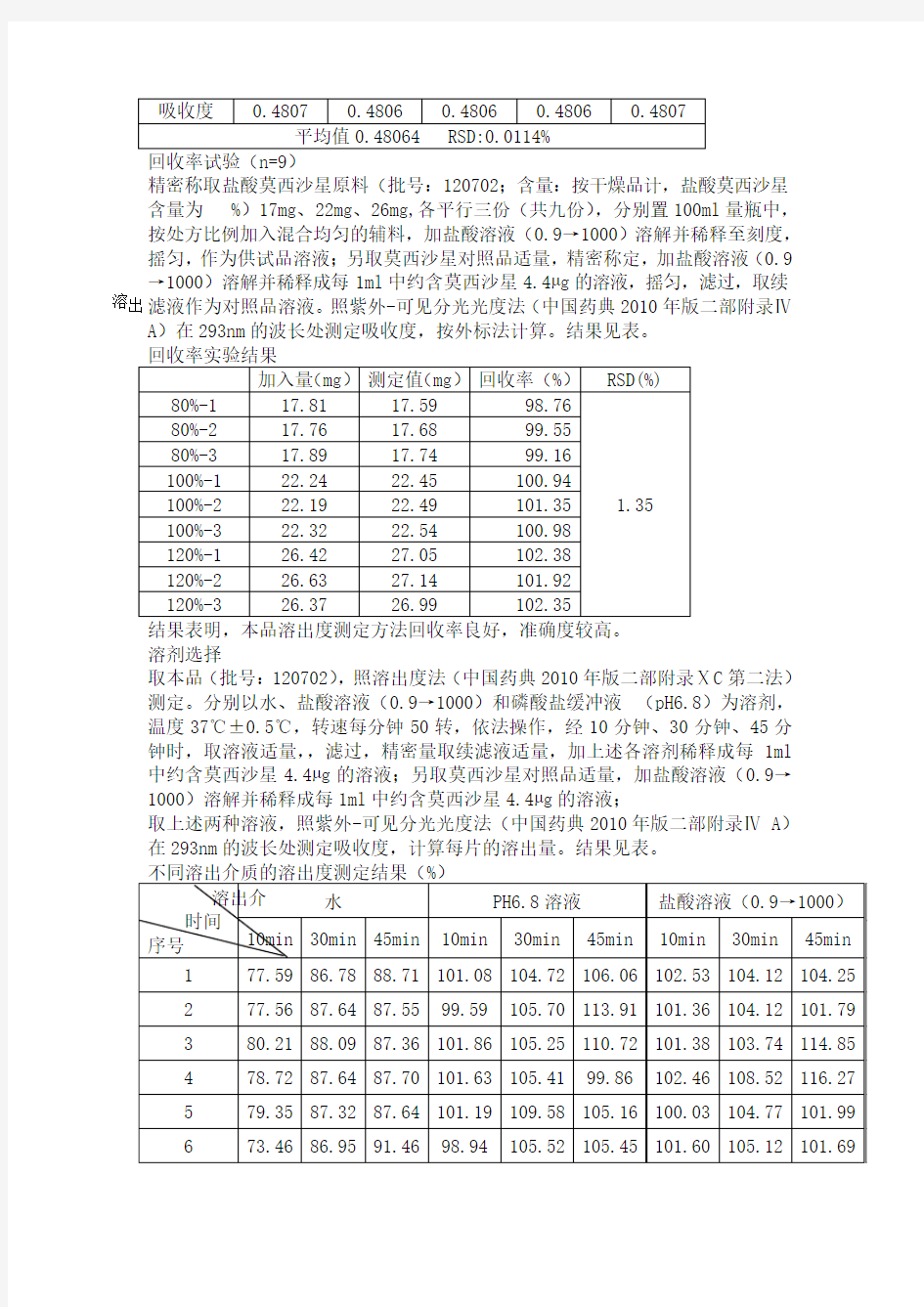
精密称取盐酸莫西沙星原料(批号:120702;含量:按干燥品计,盐酸莫西沙星含量为 %)17mg、22mg、26mg,各平行三份(共九份),分别置100ml量瓶中,按处方比例加入混合均匀的辅料,加盐酸溶液(0.9→1000)溶解并稀释至刻度,摇匀,作为供试品溶液;另取莫西沙星对照品适量,精密称定,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,摇匀,滤过,取续溶出
滤液作为对照品溶液。照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ
A)在293nm的波长处测定吸收度,按外标法计算。结果见表。
溶剂选择
取本品(批号:120702),照溶出度法(中国药典2010年版二部附录ⅩC第二法)
测定。分别以水、盐酸溶液(0.9→1000)和磷酸盐缓冲液(pH6.8)为溶剂,
温度37℃±0.5℃,转速每分钟50转,依法操作,经10分钟、30分钟、45分
钟时,取溶液适量,,滤过,精密量取续滤液适量,加上述各溶剂稀释成每1ml
中约含莫西沙星4.4μg的溶液;另取莫西沙星对照品适量,加盐酸溶液(0.9→
1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液;
取上述两种溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)
在293nm的波长处测定吸收度,计算每片的溶出量。结果见表。
上表的数据表明:本品除在水中溶出度稍差外,在盐酸溶液(0.9→1000)和磷酸盐缓冲液(pH6.8)中样品均溶出完全,无明显差别,选择用盐酸溶液(0.9→1000)作为溶出介质。
转篮法和桨法、取样时间的选择
取本品(批号:120702)6片,以盐酸溶液(0.9→1000)900ml为溶剂,温度:37℃±0.5℃,转速每分钟50转,分别照溶出度测定法第一法(转篮法)和第二法(桨法)操作,经10分钟、30分钟、45分钟时,取溶液适量,滤过,精密量取续滤液适量,加盐酸溶液(0.9→1000)稀释成每1ml中约含莫西沙星4.4μg 的溶液,作为供试品溶液;另取莫西沙星对照品适量,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,作为对照品溶液;照上述各溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在293nm的波长处测定吸收度,计算每片的溶出量。结果见表。
点的溶出结果无明显差异,为更好的控制本品质量,选择桨法,45分钟作为取样点。
溶出均一性
溶出均一性试验方法采用中国药典2010年版二部附录ⅩC第二法,随机取120702批样品36片,以盐酸溶液(0.9→1000)900ml为溶剂,温度:37℃±0.5℃,转速每分钟50转,依法操作,经45分钟时,取溶液适量,滤过,精密量取续滤液适量,加盐酸溶液(0.9→1000)稀释成每1ml中约含莫西沙星4.4μg的溶液,作为供试品溶液;另取莫西沙星对照品适量,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,作为对照品溶液;照上述各溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在293nm的波长处测定吸收度,计算每片的溶出量。36片的平均溶出量及其RSD值。结果见表。
结果表明,120702批样品的平均溶出度为,RSD为1.52%
累积溶出试验
采用中国药典2010年版二部附录ⅩC第二法,随机抽取样品6片,以盐酸溶液(0.9→1000)900ml为溶出介质,温度:37℃±0.5℃,转速每分钟50转,依法操作。经5、10、20、30、45、60分钟分别取溶出液10ml(同时补充同体积溶剂),滤过,精密量取续滤液1ml置100ml量瓶中,加盐酸溶液(0.9→1000)稀释至刻度,摇匀,作为供试品溶液;另取莫西沙星对照品适量,加盐酸溶液(0.9→1000)溶解并稀释成每1ml中约含莫西沙星4.4μg的溶液,作为对照品溶液;照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A)在293nm的波长处测定吸收度,按外标法计算各时间点的平均溶出百分数(%),以取样时间为横坐标,溶出度平均值为纵坐标,绘制累积溶出曲线。结果见表。
影响因素试验的溶出度测定 测定方法参照美国药典盐酸二甲双胍缓释片质量标准。 照释放度测定法(中国药典2010年版二部附录X D第一法),采用溶出度测定法(中国药典2010年版二部附录XC第一法蓝法)的装置,以pH6.8磷酸二氢钾缓冲液(1000ml水中加入6.8g磷酸二氢钾,用0.2N的氢氧化钠溶液调pH为6.8 ± 0.1)1000ml为溶剂,转速为每分钟100转,按溶出度测定法依法操作。分别于预定时间取溶液5ml滤过(并及时向溶出杯中补充同温度的溶剂5ml),取续滤液用释放介质稀释至适当浓度,照紫外分光光度法(中国药典2010版二部附录IV A),在232nm处测定吸光度。另精密称取盐酸二甲双胍对照品适量,用释放介质配制成约5μg/ml浓度的溶液作为对照品溶液,计算出每片的释放度。 一、溶出介质的配制 用电子天平称量磷酸二氢钾(固体)xxxg,氢氧化钠(固体)xxxg,置1000ml 烧杯中,用800ml蒸馏水溶解后,倒入10L广口瓶中,再用蒸馏水稀释至10L,配得缓冲介质。 二、对照品溶液的配制 各置于100ml容量瓶中,用溶出介质溶解并定溶至刻度;用1ml移液管各精密量取1ml至50ml容量瓶中,用溶出介质定溶至刻度。. 各样品称量值自己列出。 三、试验过程 向溶出仪6个溶出杯中各加入1000ml已配好的溶出介质,加热,待溶出杯中溶液温度达到37℃后,将6片药片同时放到6个溶出杯中后,立即开始搅拌并计时。在1h、3h、5h、7h、10h时,用10ml的注射器各取样5ml,同时向溶出杯中补加同温度溶出介质5ml。 1h、3h样品取出后,过0.45um微孔滤膜,弃去2ml初滤液,取3ml续滤液;1h样品稀释25倍后测其吸光度;3h样品稀释50倍后测其吸光度。 四、实验结果见下表 计算公式:(1)校正因子f f=(f1+f2+f3)/3 f1=C1/A1; f2=C2/A2; f3=C3/A3 C1、C2、C3:三份对照品的浓度 A1、A2、A3:三份对照品的吸光度 (2)累积释放度 result=(f*A*n*v+C1h*5+ C3h*5+…..)*v*100/m
发布日期20071130 栏目化药药物评价>>综合评价 标题浅谈溶出度检查方法的建立 作者王亚敏 部门 正文内容 审评五部王亚敏 溶出度系指药物从片剂、胶囊剂和颗粒剂等固体制剂在规定的条件下溶出的速率和程度。它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验法,是评价和控制药品制剂质量的一个重要指标,对评估制剂的批次质量、优化处方及制备工艺、保证处方工艺等变更前后产品质量的一致性有重要作用。 在中国药典附录收载的“溶出度测定法”种,对转篮法(一法)和桨法(二法)小杯法(三法)的仪器装置、测定方法和结果判定标准给予了较详细的规定。但是,如何研究和建立一个有效的溶出度检查方法,是药品研发者和生产者更加关注的问题。 本文参考拟在新版美国药典中增加的<1092>章节“溶出度检查方法的建立和验证”(The dissolution Procedure: Development and Validation),结合笔者的工作体会,重点介绍溶出度检查方法的建立相关内容。
1、了解原料药和制剂的相关理化性质。 在建立溶出度检查方法前,需首先了解原料药和制剂的相关理化性质。 对于原料药,有两方面需要了解,一是药物在不同pH条件下的溶解度,或在不同介质中的溶解度,二是药物在溶液状态下的药物的稳定性。由于溶出度检查方法要求药物在选择的介质中可以满足漏槽条件的要求,了解不同pH条件下的溶解度对介质的选择有重要意义。需要注意的是,当通过调节介质组成(如表面活性剂、pH、缓冲液等)以达到漏槽条件时,需注意评估表面活性剂、pH、缓冲液对药物溶解性和稳定性的影响。 药物pH 溶解度曲线的测定应在37±1℃下进行,测定pH1.0 7.5的水性介质中药物的溶解度。pH值测定个数需依据药物的离子化特性来决定,例如,当药物的pka为3 5时,药物的溶解度应在pH=pka, pH=pka+1, pH=pka-1, pH=1和pH=7.5处测定,pH值测定个数应可以满足准确绘制pH 溶解度曲线的需要。每个pH处溶解度数值至少测定,并根据实验结果的偏差情况适当增加测定次数。药典中收载的缓冲溶液可以用于溶解度测定,如果这些缓冲液的理化性质不适合于测定药物的溶解度,可用其他的缓冲液代替。药物加入缓冲液后应注意验证溶液的pH值。溶解度测定除传统的摇瓶法外,若有试验数据证明其他方法可测定被测药物的平衡溶解度,也可采用其他方法。缓冲溶液中药物的浓度应使用专属性好且重现性好的实验方法测定,该方法应可以将原型药物与其降解产物有效分离。 对于制剂,可能影响溶出的重要因素有制剂包衣情况、硬度、脆碎度、崩解时限、处方中增溶剂情况和其他辅料的影响。辅料有时会影响药物的吸收速度与程度,如大剂量表面活性剂(如聚山梨酯80)通常会增加药物的溶解度和加速药物的溶出。而片剂中使用比重较轻辅料量较大或使用蜡质辅料时,片剂在介质中易漂浮,选择转篮法测定结果的均一性较好。 2、溶出仪的选择 溶出仪的选择可根据制剂处方设计和制剂在体外溶出体系中的实际行为进行选择。可以
×××片溶出度试验方法学验证 1、溶出度 依据国家食品药品监督管理局国家药品标准新药转正标准第二十八册×××片溶出度试验方法、《中国药典》2010年版二部溶出度测定法(附录Ⅹ C)的有关要求,并参照文献进行本品的溶出度研究。 (1)溶出介质及介质体积的选择 溶出介质应根据制剂的特性选用水、0.01~0.1mol/L盐酸溶液或适宜的缓冲液(pH值一般不超过7.6),应临用新制并经脱气处理。对于极难溶出的品种,可加适量表面活性剂,如十二烷基硫酸钠(0.5%以下),如确需使用有机溶剂,可加适量,如异丙醇、乙醇等(通常浓度在5%以下),但应有依据,并尽量选用低浓度。 溶出介质的体积一般应符合漏槽条件。 ×××在水中微溶,且本品为小规格品种(规格为2.5mg),根据以上溶出介质选择的原则及已有国家标准的方法,选择已有国家标准中采用的溶出介质及体积:0.1mol/L盐酸溶液〔盐酸溶液(9→1000)〕200ml。 (2)溶出方法及其转速的选择 方法的选择一般可参照下列原则: ①对于非崩解型药物,宜采用转篮法。 ②对于崩解型药物,在进行转篮法的整个试验过程中,确保转篮网孔的通透性尤为重要,对于处方中主药或辅料(如胶性物质)影响转篮通透性的固体制剂,一般应采用桨法。 ③制剂中含有难以溶解、扩散的成分,一般应采用桨法。 ④对飘浮于液面的制剂,一般应选用转篮法。如辅料堵塞网孔则选用桨法,将供试品放入沉降篮中,并在正文中加以规定。采用小杯法时不能使用沉降篮。 ⑤小杯法主要用于在转篮法和桨法条件下,溶出液的浓度过稀,即使采用较灵敏的方法仍难以进行定量测定的品种。 转速选择的原则:在质量研究的基础上,尽量选择低转速,转篮法推荐100转/分,最低不得低于50转/分;桨法推荐50转/分,最高不超过75转/分;小杯
实验室溶出度测定法规程
实验室溶出度测定法规程 目的:建立溶出度测定法标准操作规程。 适用范围:溶出度测定。 责任:质检员实施本操作规程,检验室主任负责监督本规程正确执行。 程序: 1.简述 1.1溶出度(中国药典2000年版二部附录X C)是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出的量。 1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。 1.4中国药典2000年版收载三种测定方法,第一法转篮法第二法桨法及第三法小杯法。 1.5凡检查溶出度的制剂,不再进行崩解时限的检查。 2.仪器与用具 2.1溶出度仪 2.1.1仪器的组成溶出度仪主要由电动机、恒温水 浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详见中国药典2000年版二部附录X C。 2.1.2仪器的装置与使用按仪器使用说明书及中国药
典的规定进行安装与使用。 2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 2.1. 3.1溶出度校正片分崩解型和非崩解型两种,崩解型为泼尼松片,非崩解型为水杨酸片。目前国内仅有非崩解型校正片。 2.1. 3.2校正前,应先调式所用仪器。 2.1. 3.3溶剂:磷酸盐缓冲液(PH7.4)。配制方法见中国药典2000年版二部附录XV D,要求PH值为7.40±0.05,临用前脱气。 2.1. 3.4对照品溶液的制备取溶出度校正用水杨酸片1片,精密称定,置乳体中,研细,精密称取适量(约相当于水杨酸10mg),置100ml量瓶中,加乙醇1ml,摇匀,加溶剂适量,经超声处理30分钟,使水杨酸溶解,加溶剂到刻度,摇匀,经滤纸(不宜使用滤膜)滤过,取续滤液为对照品溶液。(对照应做2份平行试验) 2.1. 3.5校正溶液的制备取溶剂各900ml,分别注入每个操作容器中,温度保持在37±0.5℃,按规定(桨法为50转/分钟;篮法为100转/分钟)调整转速。取溶出度校正用水杨酸片6片,分别精密称定,分置6个容器中,自药片接触溶出介质时,开始计时,并分别在10、15、20、25和30分钟时取样(连续取样不停机),每次抽取2ml(及时补充溶剂2ml),各自经滤纸滤过(六个小漏斗和六张滤纸,连续使用,每次滤过后,漏斗底部应无液体存在),取续滤液为校正溶液。 2.1. 3.6测定法精密吸取对照品溶液及校正溶液各
附件1 普通口服固体制剂溶出度试验 技术指导原则 一、前言 本指导原则适用于普通口服固体制剂,包括以下内容:(1)溶出度试验的一般要求;(2)根据生物药剂学特性建立溶出度标准的方法;(3)溶出曲线比较的统计学方法;(4)体内生物等效性试验豁免(即采用体外溶出度试验代替体内生物等效性试验)的一般考虑。 本指导原则还针对药品的处方工艺在批准后发生变更时,如何通过溶出度试验确认药品质量和疗效的一致性提出了建议。附录对溶出度试验的方法学、仪器和操作条件进行了概述。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透。由于药物的溶出和溶解对吸收具有重要影响,因此,体外溶出度试验有可能预测其体内行为。基于上述考虑,建立普通口服固体制剂(如片剂和胶囊)体外溶出度试验方法,有下列作用: 1.评价药品批间质量的一致性; 2.指导新制剂的研发;
3.在药品发生某些变更后(如处方、生产工艺、生产场所变更和生产工艺放大),确认药品质量和疗效的一致性。 在药品批准过程中确定溶出度标准时,应考虑到药物的溶解性、渗透性、溶出行为及药代动力学特性等因素,以保证药品批间质量的一致性、变更以及工艺放大前后药品质量的一致性。 对于新药申请,应提供关键临床试验和/或生物利用度试验用样品以及其他人体试验用样品的体外溶出度数据。对于仿制药申请,应在溶出曲线研究的基础上制定溶出度标准。无论是新药还是仿制药申请,均应根据可接受的临床试验用样品、生物利用度和/或生物等效性试验用样品的溶出度结果,制定溶出度标准。 三、生物药剂学分类系统 根据药物的溶解性和渗透性,推荐以下生物药剂学分类系统(BCS)(Amidon 1995): 1类:高溶解性–高渗透性药物 2类:低溶解性–高渗透性药物 3类:高溶解性–低渗透性药物 4类:低溶解性–低渗透性药物 上述分类原则可作为制定体外溶出度质量标准的依据,也可用于预测能否建立良好的体内-体外相关性(IVIVC)。在37±1℃下,测定最高剂量单位的药物在250mL pH值介于1.0和8.0之间的溶出介质中的浓度,当药物的最高剂量除以以上介质中的药物浓度小于或等于250mL时,可认为是高溶解性药物。一般情
溶出度总结 一、溶出度方法的确立 1、溶出方法的选择 (1)篮法(B)/浆法(P),不提首选浆法或蓝法 非崩解型药物(B) 崩解型药物制剂中含有难以溶解、扩散的成分(P) 主药或辅料为一定胶性物质(P) 悬浮的制剂(B),如辅料易堵塞网孔(P,使用沉降篮) (2)小杯法:≥500ml浓度过低,较灵敏的方法仍难以进行定量测定(不能使用沉降篮,测定不能再稀释测定)。 2、溶出介质的选择 (1)水:不提以水为主(pH 值无法控制,在试验过程中易发生改变,适合非pH 依赖释药)(2)人工胃液(0.01~0.1mol/L盐酸溶液, 必要时可加胃蛋白酶)(3)人工肠液(必要时可加胰蛋白酶) (4)其他缓冲液(pH值一般不超过7.6)三羟甲基氨基甲烷(Tris):缓冲范围pH7.0~9.5,低离子强度(二氟尼柳胶囊) (5)其他: 低浓度表面活性剂; 醇溶液(一般<5%)人体生理pH值在胃内为1~3.5,小肠内约为7,结肠内约为7.5 (6)表面活性剂----尽量避免使用,种类和浓度需通过多个试验来验证。?FDA 溶出度指导原则:对于难溶性药物不提倡使用有机溶剂,推荐SDS,但必须证明表面活性剂的选择和用量的合理性。即应考察表面活性剂对药物的增溶量,以确定最少且最佳的使用浓度。 采用阳离子表面活性剂/非离子表面活性剂。十二烷基硫酸钠(Sodium laurylsulfate SLS或SDS) —纯度;pH≦2.5聚山梨酯(吐温)20-80 (Tween20-80 ) —茴三硫片/吉非替尼片/盐酸雷洛昔芬片等溴化十六烷基三甲胺—粘度大月桂基二甲基氧化铵—替代SDS 用于胶囊剂(可以与酶配伍) 3、溶出介质体积的选择 使药物符合漏槽条件小规格品种一般不提倡将2粒/片投入1个溶出杯中来满足测定的灵敏度需要。常用:大杯法:500 ~1000ml ,900ml为最普遍小杯
1.目的 建立溶出度测定法操作规程。 2.适用范围 本规程适用于溶出度测定法。 3.编制依据 《药品生产质量管理规范(1998年修订)》国家药品监督管理局(1999)4.责任 QC主管、QC质检员对本规程的实施负责。 5.正文 5.1简述 5.1.1溶出度(中国药典2010年版二部附录X C)是指药物从片剂\胶囊剂或颗粒剂等固体制剂在规定条件中溶出速率和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 5.1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出量。 5.1.3中国药典2010年版收载三种测定方法,第一法转篮法,第二法桨法及第三法小杯法。 5.1.4除另有规定外,凡检查溶出度的制剂,不再进行崩解时限的检查。 5.2仪器与用具 5.2.1溶出度仪 5.2.1.1仪器原组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详见中国药典2010年版二部附录X C。 5.2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。5.2.1.3仪器的校正为使药物的溶出度测定结果准确、可靠,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 5.2.1.4仪器的调试 5.2.1.4.1检查仪器水平及转动轴的垂直度与偏心度(使用水平以检查仪器是否处于水平状态;转轴的垂直程度应与容器中心线相吻合,用直角三角板检查转动轴与溶出杯平
溶出度总结 一、溶出度方法得确立 1、溶出方法得选择 (1)篮法(B)/浆法(P),不提首选浆法或蓝法 非崩解型药物(B) 崩解型药物制剂中含有难以溶解、扩散得成分(P) 主药或辅料为一定胶性物质(P) 悬浮得制剂(B),如辅料易堵塞网孔(P,使用沉降篮) (2)小杯法:≥500ml浓度过低,较灵敏得方法仍难以进行定量测定(不能使用沉降篮,测定不能再稀释测定)。 2、溶出介质得选择 (1)水:不提以水为主(pH 值无法控制,在试验过程中易发生改变,适合非pH 依赖释药)(2)人工胃液(0、01~0、1mol/L盐酸溶液, 必要时可加胃蛋白酶)(3)人工肠液(必要时可加胰蛋白酶) (4)其她缓冲液(pH值一般不超过7、6)三羟甲基氨基甲烷(Tris):缓冲范围pH7、0~9、5,低离子强度(二氟尼柳胶囊) (5)其她: 低浓度表面活性剂; 醇溶液(一般<5%)人体生理pH值在胃内为1~3、5,小肠内约为7,结肠内约为7、5 (6)表面活性剂----尽量避免使用,种类与浓度需通过多个试验来验证。?FDA 溶出度指导原则:对于难溶性药物不提倡使用有机溶剂,推荐SDS,但必须证明表面活性剂得选择与用量得合理性。即应考察表面活性剂对药物得增溶量,以确定最少且最佳得使用浓度。 采用阳离子表面活性剂/非离子表面活性剂。十二烷基硫酸钠(Sodium laurylsulfate SLS或SDS) —纯度;pH≮2、5聚山梨酯(吐温)20-80 (Tween20-80 ) —茴三硫片/吉非替尼片/盐酸雷洛昔芬片等溴化十六烷基三甲胺—粘度大月桂基二甲基氧化铵—替代SDS 用于胶囊剂(可以与酶配伍) 3、溶出介质体积得选择 使药物符合漏槽条件小规格品种一般不提倡将2粒/片投入1个溶出杯中来满足测定得灵敏度需要。常用:大杯法:500 ~1000ml ,900ml为最普遍小杯
溶出度测定法标准操作规程 目的:建立溶出度测定法标准操作规程。 适用范围:溶出度测定。 责任:质检员实施本操作规程,检验室主任负责监督本规程正确执行。 程序: 1.简述 1.1溶出度(中国药典2000年版二部附录X C)是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标,是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或烧杯)中,在37.0±0.5℃恒温下,在规定的转速、溶剂中依法操作,在规定的时间内测定其溶出的量。 1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。 1.4中国药典2000年版收载三种测定方法,第一法转篮法第二法桨法及第三法小杯法。 1.5凡检查溶出度的制剂,不再进行崩解时限的检查。 2.仪器与用具 2.1溶出度仪 2.1.1仪器的组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成,详 见中国药典2000年版二部附录X C。 2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。 2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性,应对新安装的溶出度仪采用溶出度校正片进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 2.1. 3.1溶出度校正片分崩解型和非崩解型两种,崩解型为泼尼松片,非崩解型为水杨酸片。目前国内仅有非崩解型校正片。 2.1. 3.2校正前,应先调式所用仪器。 2.1. 3.3溶剂:磷酸盐缓冲液(PH7.4)。配制方法见中国药典2000年版二部附录XV D,要求PH值为7.40±0.05,临用前脱气。 2.1. 3.4对照品溶液的制备取溶出度校正用水杨酸片1片,精密称定,置乳体中,研细,精密称取适量
溶出度系指药物从片剂或胶囊剂等固体制剂在规定溶剂中溶出的速度和程度。凡检查溶出度的制剂,不再进行崩解时限的检查。第一法仪器装置(1)转篮分篮体与篮轴两部分,均为不锈钢金属材料制成。篮体A由不锈钢丝网(丝径为0.254mm,孔径0.425mm)焊接而成,呈圆柱形,内径为22.2±1.0mm,上下两端都有金属边缘。篮轴B的直径为9.4~10.1mm,轴的末端连一金属片,作为转篮的盖;盖上有通气孔(孔径 2.0mm);盖边系两层,上层外径与转篮外径同,下层直径与转篮内径同;盖上的三个弹簧片与中心呈120°角。转篮旋转时摆动幅度不得超过±1.0mm。(2)操作容器为1000ml的圆底烧杯,内径为98~106mm,高160~175mm;烧杯上有一有机玻璃盖,盖上有2孔,中心孔为篮轴的位置,另一孔供取样或测温度用。为使操作容器保持恒温,应外套水浴;水浴的温度应能使容器内溶剂的温度保持在37±0.5℃。转篮底部离烧杯底部的距离为25±2mm。(3)电动机与篮轴相连,转速可任意调节在每分钟50~200转,稳速误差不超过±4%。运转时整套装置应保持平稳,不得晃动或振动。(4)仪器应装有6套操作装置,可一次测定6份供试品。取样点位置应在转篮上端距液面中间,离烧杯壁10mm处。测定法除另有规定外,量取经脱气处理的溶剂900ml,注入每个操作容器内,加温使溶剂温度保持在37±0.5℃,调整转速使其稳定。取供试品6片(个),分别投入6个转篮内,将转篮降入容器中,立即开始计时,除另有规定外,至45分钟时,在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成。取滤液,照各药品项下规定的方法测定,算出每片(个)的溶出量。结果判断6片(个)中每片(个)的溶出量,按标示含量计算,均应不低于规定限度(Q);除另有规定外,限度(Q)为标示含量的70%。如6片(个)中仅有1~2片(个)低于规定限度,但不低于Q-10%,且其平均溶出量不低于规定限度时,仍可判为符合规定。如6片(个)中有1片(个)低于Q-10%,应另取6片(个)复试;初、复试的12片(个)中仅有1~2片(个)低于Q-10%,且其平均溶出量不低于规定限度时,亦可判为符合规定。供试品的取用量如为2片(个)或2片(个)以上时,算出每片(个)的溶出量,均不得低于规定限度(Q);不再复试。
(1092)溶出度试验的开发和验证【中英文对照版】 INTRODUCTION 前言 Purpose 目的 The Dissolution Procedure: Developmentand Validation <1092> provides a comprehensive approach covering items to considerfor developing and validating dissolution procedures and the accompanyinganalytical procedures. It addresses the use of automation throughout the testand provides guidance and criteria for validation. It also addresses thetreatment of the data generated and the interpretation of acceptance criteriafor immediate- and modified-release solid oral dosage forms. 溶出实验:开发和验证(1092)指导原则提供了在溶出度方法开发和验证过程中以及采用相应分析方法时需要考虑的因素。本指导原则贯穿溶出度实验的全部过程,并对方法提供了指导和验证标准。同时它还涉及对普通制剂和缓释制剂所生成的数据和接受标准进行说明。 Scope 范围 Chapter <1092> addresses the development andvalidation of dissolution procedures, with a focus on solid oral dosage forms.Many of the concepts presented, however, may be applicable to other dosageforms and routes of administration. General recommendations are given with theunderstanding that modifications of the apparatus and procedures as given in USP general chapters need to be justified. <1092>章节讨论了溶出度实验的开发和验证,重点是口服固体制剂。所提出的许多概念也可能适用于其他剂型和给药途径。关于设备和方法的修改部分在USP通则中给出了合理的说明。 The organization of <1092> follows the sequence of actions often performed inthe development and validation of a dissolution test. The sections appear inthe following sequence. 在进行溶解度实验的开发和验证时,常遵循指导原则<1092>,具体内容如下:1. PRELIMINARY ASSESSMENT (FOR EARLY STAGES OF PRODUCTDEVELOPMENT/DISSOLUTION METHOD DEVELOPMENT) 1.前期评估(对产品开发以及溶出度方法开发的前期研究评估) 1.1 Performing Filter Compatibility 1.1滤膜相容性研究 1.2 Determining Solubility and Stability of DrugSubstance in Various Media 1.2原料药在不同溶出介质中溶解度测定和稳定性研究
溶出度测定标准操作规程 1 简述 1.1 溶出度(中国药典2005年版二部附录Ⅹ C)系指测定药物从片剂、胶囊剂或颗粒剂等固体制剂在规定条件下溶出的速率和程度。它是评价药物口服固体制剂质量的一个指标,是一种摸拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2 溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮(或溶出杯)中,在37.0℃±0.5℃恒温下,在规定的转速、溶出介质中依法操作,在规定的时间内取样并测定其溶出量。 1.3 中国药典2005年版收载三种测定方法,第一法为转篮法,第二法为桨法及第三法为小杯法。 1.4 除另有规定外,凡检查溶出度的制剂,不再进行崩解时限的检查。 2 仪器与用具 2.1 溶出度仪 2.1.1 仪器的组成溶出度仪由电动机、恒温水浴、篮体、篮轴、搅拌桨、溶出杯及杯盖等组成,详见中国药典2005年版二部附录ⅩC。 2.1.2 仪器的装置与使用按仪器使用说明书及中国药典对溶出度的规定进行安装与使用。 2.1.3 仪器的校正为使药物的溶出度测定结果准确、可靠,应
对新安装的溶出度仪按溶出度校正片说明书进行校正,对已使用过的仪器也应定期(或在出现异常情况时)进行校正。 2.1.4 仪器的调试 2.1.4.1 检查仪器水平及转动轴的垂直度与偏心度(使用水平仪检查仪器是否处于水平状态;转轴的垂直程度应与容器中心线相吻合,用直角三角板检查转动轴与溶出杯平面的垂直度;检查转篮旋转时与溶出杯的垂直轴在任一点的偏离均不得大于2mm,检查转篮旋转时摆动幅度不得偏离轴心的±1.0mm;或检查桨杆旋转时与溶出杯的垂直轴在任一点的偏差均不得大于2mm;或检查搅拌桨旋转使A、B 两点的摆动幅度不得大于0.5mm。 2.1.4.2 篮轴运转使整套装置应保持平稳,均不能产生明显的晃动或振动(包括仪器装置所放置的环境)。 2.1.4.3 转速与允差范围检测仪器的实际转速与其仪器的电子显示的数据是否一致,稳速误差不得超过±4%。 2.2 取样器注射器(5、10、15、20ml等适合的注射器)及取样针头。 2.3 过滤器一般常用滤头及滤膜(不同规格,孔径不得大于0.8μm)。 3 溶出度测定前的准备 3.1 测定前,应对仪器装置进行必要的调试,第一法使转篮底部距溶出杯的内底部25mm±2mm;第二法使桨叶底部距溶出杯的内底部25mm±2mm;第三法使桨叶底部距溶出杯的内底部15m m±2mm。
化学药品普通口服固体制剂溶出度方法验证易忽视的几个问题 审评四部审评八室郑国钢 溶出度系指药物从片剂或胶囊剂等固体制剂在规定的溶出介质中溶出的速度和程度,是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。它是评价药物制剂质量的一个重要指标。 一个完整的溶出度方法验证主要包括以下内容:(1)溶出介质及介质体积的选择;(2)溶出方法(转篮法与桨法)及其转速的选择;(3)溶出量测定方法的验证,(4)溶出度均一性试验(批内)、重现性试验(批间)等。审评中发现提供溶出度方法验证资料往往不全,应引起申报单位注意。 (一)溶出度测定方法的选择 溶出度测定方法的选择包括溶出介质及介质体积的选择、溶出方法(转篮法与桨法)及其转速的选择。根据《化学药物质量标准建立的规范化过程技术指导原则》,溶出介质通常采用水、0.1mol/L盐酸溶液、缓冲液(pH值3~8为主)。对在上述溶出介质中均不能完全溶解的难溶性药物,可加入适量的表面活性剂,如十二烷基硫酸钠等。检查方法转篮法以100转/分钟为主;桨法以50转/分钟为主。 应该注意的是(1)溶出介质的体积需使药物符合漏槽条件,大杯法(第一、二法)常用体积为500~1000ml,小杯法(第三法)常用体积为100~250ml。部分品种为满足在溶出量测定时药物浓度的需要,可采用低于上述限度范围的溶剂。(2)介质、方法、转速的选择一般根据溶出曲线测定结果确定。部分资料简单地通过比较主药在各溶剂中的溶解度来选择溶出介质,我们认为相同的溶剂可能会导致对不同制剂溶出行为的差异,且工艺的选择、辅料的加入能改变主药在不同溶剂中的溶解行为,故仅考虑溶解度是不适合的;部分资料根据单点测定结果进行方法和转速选择,如盐酸左旋多巴甲酯片申报资料中采用篮法100rpm和桨法75rpm比较,结果45min溶出均大于95%,故选择桨法75rpm测定溶出度,单点测定不能很好区分不同处方和生产工艺的溶出情况,也影响溶出拐点的确定,故不合适;考虑今后大生产工艺,申报单位确定溶出度检查方法中常采用高转速或延长取样时间,取样时间与溶出曲线的拐点位置相距较远,导致溶出度测定区分能力不明显,溶出度取样时间常选择溶出曲线的拐点处后推10~20分钟,如果时间较长或太短,可通过适当提高或减低转速等手段重新测定溶出曲线。(3)如是仿制已有国家标准的药品,则
陕西香菊药业集团有限公司GMP管理文件 1.目的:本标准规定了溶出度的测定方法和操作要求。 2.范围:本公司检品的溶出度的测定。 3.责任人:QC检验员、QC主任。 4. 引用标准:《中华人民共和国药典》2015版四部 5.内容: 5.1.仪器:溶出度仪、注射器及取样针头、过滤器、滤膜。 5.2.简述;溶出度系指药物从片剂、胶囊剂或颗粒剂等固体制剂在规定条件下溶出的速率和程度。凡检查溶出度的制剂,不再进行崩解时限的检查。 5.3.操作方法 5.3.1.仪器装置: 5.3.1.1.搅拌桨:由不锈钢材料制成,搅拌桨的下端及桨叶部分可使用涂有合适的惰性物质的材料, 桨杆旋转时与溶出杯的垂直轴在任一点的偏差均不得大于2mm, 搅拌桨旋转时两点的摆动幅度不得超过0.5mm。 5.3.1.2溶出杯: 由硬质玻璃或其他惰性材料制成的透明或棕色的、底部为半球形的1000ml(小杯250ml)杯状容器,内径为102mm±4mm(小杯62mm±3),高为168mm ±8mm(小杯126mm+_6mm)。 5.3.2.测定法:测定前应对仪器装置进行必要的调试,使浆叶底部距溶出杯内底部25mm±2mm,除另有规定外,分别量取经脱气处理的溶出介质900ml,置各溶出杯内,加温,待溶出介质温度恒定在37℃±0.5℃,按照各品种项下的规定调节电动机转速,待其平稳后,取供试品6片(袋、粒),分别投入6个溶出杯内,(除另有规定外,如片剂或胶囊剂浮于液面,应先装入沉降篮内),自供试品接触介质时起,立即计时,至规定的取样时间,吸取溶出液适量,(取样位置应在桨叶顶端至液面的中点,距溶出杯内壁不小于
10mm,小杯法不小于6mm处取样)立即用适当的微孔滤膜滤过,自取样至滤过应在30秒内完成。取澄清滤液,照各品种项下规定的方法测定,计算每片(袋、粒)的溶出量。 5.3.3.结果判定符合下述条件之一者,可判为符合规定: 5.3.3.1. 6粒中,每粒的溶出量按标示量计算均不得低于规定限度(Q)。 5.3.3.2. 6粒中,如有1-2粒低于Q,但不低于Q-10%且其平均溶出量不低于Q。 5.3.3.3. 6粒中有1-2粒低于Q,其中仅有一粒低于Q-10%,但不低于Q-20%,且其平均溶出量不低于Q时,应另取6粒复试。复试中,初复试的12 粒中有1-3粒(片)低于Q,其中仅有1粒低于Q-10%,但不低于Q-20%,且其平均溶出量不低于Q。 5.3.3.4. 10%,20%指相对标示量的百分率。 5.3.4.溶出条件及注意事项; 5.3.4.1.溶出度仪的校正;除仪器的各项机械性能应符合上述规定外,还应用校正片校正,按照校正说明书操作,试验结果应符合校正的规定。 5.3.4.2.溶出介质应使用各品种项下规定的溶出介质,并应新鲜配置和经脱气处理(溶解的气体在试验的过程中可能形成气泡,从而影响试验结果,因此溶解的气体应在试验前除去。脱气方法:取溶出介质,采用超声除气方法)如果溶出介质为缓冲液,调节pH值至规定的pH值±0.05之内。 5.3.4.3.取样时间应按照各品种项下规定的取样时间,自6杯中完成取样的时间应在1分钟内。 5.3.4.4.如胶囊壳对分析有干扰,应取不少于6粒胶囊尽可能完全地除尽内容物,置同一溶出杯内,用该品种项下规定体积的容出介质溶解空胶囊壳,并按该品种项下的分析方法测定每个空胶囊的空白值,作必要的校正,如校正值大于标示量的25%,试验无效。如校正值不大于标示量的2%可忽略不计。 5.3.4.5.除另有规定外,取样时间为45分钟,限度为标示量的70%。 5.3.4. 6.测定时,除另有规定外每个溶出杯中只允许投入供试品1片(袋、粒)。
溶出度(释放度)检测方法建立及验证标准操作规程 1.目的 为保证检测工作的可靠性和可重现性,在未知样品的检测前必须对检测方法进行验证以证明所采用的检测方法适合于相应的检测要求。 2.范围 建立药品质量标准时、药品生产工艺变更时、制剂组分发生变更时、原分析方法修订时均应进行溶出度或释放度测定的方法学的验证。 3.责任人 检测员、项目负责人、各级项目经理:要求系统、全面验证含量测定方法并记录整理验证数据。 4.程序 4.1 验证内容(以下为溶出度验证方法,释放度具体详见化学药物口服缓释制剂药学研究技术指导原则。) 溶出度系指药物从片剂或胶囊剂等固体制剂在规定的溶出介质中溶出的速度和程度,是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。它是评价药物制剂质量的一个重要指标。 一个完整的溶出度方法验证主要包括以下内容:(1)溶出介质及介质体积的选择;(2)溶出方法(转篮法与桨法)及其转速的选择;(3)溶出量测定方法的验证,(4)溶出度均一性试验(批内)、重现性试验(批间)等。 4.2 验证方法 (一)溶出度测定方法的选择 溶出度测定方法的选择包括溶出介质及介质体积的选择、溶出方法(转篮法与桨法)及其转速的选择。根据《化学药物质量标准建立的规范化过程技术指导原则》,溶出介质通常采用水、0.1mol/L盐酸溶液、缓冲液(pH值3~8为主)。对在上述溶出介质中均不能完全溶解的难溶性药物,可加入适量的表面活性剂,如十二烷基硫酸钠等。检查方法转篮法以100转/分钟为主;桨法以50转/分钟为主。 应该注意的是(1)溶出介质的体积需使药物符合漏槽条件,大杯法(第一、二法)常用体积为500~1000ml,小杯法(第三法)常用体积为100~250ml。
溶出度试验影响因素及溶出方法验证 1.溶出速率的定义 Noyes-whitney 方程:dW/dt=kS(Csat-Csol) 试中:dW/dt-溶出速率 K-溶出常数 S-固体的表面积 保持溶出介质的体积至少是饱和溶液体积的3倍。则基本可以满足漏槽条件。 2.表面积的影响 非崩解型固体和崩解型固体溶出试验中表面积S 随时间t 的变化 s 表面积
溶出度仪的影响因素 1.晃动的影响 A.与TIR值小于2.0mm的结果相比,晃动偏差TIR值为1.0~2.0mm,水杨 酸片和泼尼松片的溶出量分别增加约5%。 B.溶出度仪设计中需注意两个因素1.要求转轴必须垂直;2.转轴应有两个固 定点,仪器顶部到转轴卡盘的距离至少应不低于卡盘至转篮或桨叶的距离。 2.转轴的直线度 a.转轴的直线度是控制晃动的关键仪器指标,应确保其直线度 b.溶出度仪的涉及要求桨叶或篮体的顶端距卡盘的距离至少要6英寸 (15.2cm)。 3.其它搅拌装置的变动因素 A.篮杆和桨杆是精密部件,使用时应小心,这些精密仪器在实验室的抽屉中 存放时,会破坏不锈钢桨表面的。引起弯曲和变性。应有适当的支架供转轴存放。 B.桨叶应没有锐角。尖锐部分会引起涡流而不是层流。取用或放置篮时只能 接触篮的上部边框,随着时间的增加,特别是在酸性介质时,筛网的孔径会有变化。可用放大镜检,必要时需更换篮。 4.振动 4.1振动的来源 A实验室中能产生振动的仪器,包括通风橱和离心机,空调,风扇,离心机等。 B.人员走动,关门,开门。 C.早期水浴加热和溶出仪连在一起,目前的溶出度仪设计都采用外置循环泵与水浴连接。 4.2应将盛有溶出介质的溶出杯的各种来源的振动水平降低到0.1mil。 5.搅拌装置的准直度 转轴的轴线与溶出杯的中心轴线间的偏离和倾斜对溶出介质流体动力学影
GMP管理文件 一、引用标准:中华人民共和国S药典(2005年版)一部。 二、适用范围:适用于片剂溶出度的检查。 三、目的:本标准规定了片剂溶出度检查法标准操作规程。 四、责任者:质检人员。 五、正文: 1、简述释放度系指药物从缓释制剂、控释制剂及肠溶制剂等规定 条件下释放的速率和程度。 缓释、控释、肠释制剂的分类照缓释、控释和迟释制剂指导原则的规定。 2、仪器与用具溶出度检测仪 3、操作方法 第一法照溶出度测定法项下进行,但至少采用三个时间取样,在规定取样时间点,吸取溶液适量,立即经不大于0.8um微孔滤膜滤过,自取样至滤过应在30秒内完成,并及时补充所耗的溶剂。 取滤液,照各品种项下规定的方法测定,计算每片(个)的释放量。
结果判定除另有规定外,符合下述条件这一者,可判定为符合规定; (1)6片(粒)中,每片(粒)在每个时间点测得的释放量按标示量计算,均未超出规定范围; (2)6片(粒)中,在每个时间点测得的释放量,如有1~2片(粒)超出规定范围,但未超出规定范围的10%,且在每个时间点测得的平均释放量未超出规定 范围; (3)6片(粒)中,在每个时间点测得的释放量,如有1~2片(粒)超出范围,其中仅有1片(粒)超出规定范围的10%,但未超出规定范围的20%。且其平均释放量未超出规定范围,应另取6片(粒)复试;初、复试的12片(粒)中,在每个时间点测得的释放量,如有1~3片(粒)超出规定范围,其中仅有1片(粒)超出规定范围的10%,但未超出规定范围的20%,且其平均释量未超出规定范围。 以上结果判断中所示超出规定范围的10%、20%是指相对于标示量的百分率(%),其中超出规定范围10%是指:每个时间点测得的释放量不低于低限的-10%,或不超过高限的+10%;每个时间点测得的释放量应包括最终时间则得的释放量。 第二法 用于肠溶制剂
溶出度与累积溶出度的总结 .概况 溶出度也称溶出速率,是指在规定的溶剂和条件下,药物从片剂、胶囊剂、颗粒剂等固体制剂中溶出的速度和程度。药物溶出度检查是评价制剂品质和工艺水平的一种有效手段,可以再一定的程度上反映主要的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的差异,也是评价制剂活性成分生物利用度和制剂均匀度的一种有效标准。 .溶出度与生物利用度的关系 生物利用的如果通过体内试验和临床研究去评价,费时、费钱、费精力。因此,只能借助于体外溶出度实验的方法来检验和控制产品的质量,现在的制剂水平尚达不到溶出试验结果与体内完全一致,而只能有一定得相关性,溶出度虽非必然与体内生物利用度相关,但多数情况下是相关的,溶出速率是限时因素,溶出试验被看作是介于生物等效性和药品质量控制二者之间的一项较有利的措施,它是以体外实验法代替动物试验的一种方法,溶出度与生物利用度密切相关,而溶出度的体外实验较生物利用度简单易行。 .溶出度研究试验主要包括的内容 1)溶出介质的选择; 2)溶出介质体积的选择; 3)溶出方法的选择; 4)转速的选择; 5)溶出测定方法的验证; 6)溶出度均一性试验; 7)重现性试验等; .体外溶出度评价方法和统计分析方法 溶出度的评价方法常用有种方法:对数曲线法、机率单位法、指数模式法、法、法。
对实验数据的统计分析方法有种方法:回归分析法、方差分析法、相似因子法、多变因子法、法、‘法。 .累积溶出度 (一)假如,取样测定溶出度,再补液,而后补的溶介不含主药的,所以下次测定的溶出度值不是真实的值,固在测定下一次的溶出量时,需要加上上次取样的量。这就是累积溶出度与普通溶出度的区别。 (二)累积溶出量公式:下面介绍两种方法 ①累积溶出量溶出的总物质量+投入量X 溶出的总物质量{当前取样点介质浓度X 介质体积(之前取样点介质浓度X取样量)} 例:测定一药物(规格片)的溶出度,分别在、、、、取样,每个取样点取样,而后补液(最后一次不用补液),转蓝法,溶介,六片测定。 所以:第一点溶出度所取样品测定的浓度(换算成)X± X; 第二个点的累积溶出度{所取样品测定的浓度(换算成)XX }十X ; 第三个点的累积溶出度{所取样品测定的浓度(换算成)XX X }十X ; 第四个点的累积溶出度{所取样品测定的浓度(换算成)XX X X }十X ; 第五个点的累积溶出度{所取样品测定的浓度(换算成)XX ② (()) 为第次经校正后测定的相对百分溶出度,’为第次实际测定的相对百分溶出度, 为溶出介质总体积,为每次取样后所补充的体积数。 例:每次取样,再补充,溶出介质总体积,第、、、取样测定,测的溶出度分别为、、,则累积溶出为:
实验室溶出度测定法规程 实验室溶出度测定法规程目的建立溶出度测定法标准操作规程。适用范围溶出度测定。责任质检员实施本操作规程检验室主任负责监督本规程正确执行。程序 1.简述 1.1溶出度中国药典2000年版二部附录X C是指药物从片剂或胶囊剂等口服固体制剂在规定溶剂中溶出的速度和程度。它是评价药物口服固体制剂质量的一个指标是一种模拟口服固体制剂在胃肠道中崩解和溶出的体外简易试验方法。 1.2溶出度测定法是将某种固体制剂的一定量分别置于溶出度仪的转篮或烧杯中在37.0?0.5?恒温下在规定的转速、溶剂中依法操作在规定的时间内测定其溶出的量。 1.3本方法适用于片剂、胶囊剂及颗粒剂的测定。 1.4中国药典2000年版收载三种测定方法第一法转篮法第二法桨法及第三法小杯法。 1.5凡检查溶出度的制剂不再进行崩解时限的检查。 2.仪器与用具 2.1溶出度仪 2.1.1仪器的组成溶出度仪主要由电动机、恒温水浴、篮体、篮轴、搅拌桨、圆底烧杯及杯盖组成详见中国药典2000年版二部附录X C。 2.1.2仪器的装置与使用按仪器使用说明书及中国药典的规定进行安装与使用。 2.1.3仪器的校正为使同一药物的溶出度测定得到良好的再现性应对新安装的溶出度仪采用溶出度校正片进行校正对已使用过的仪器也应定期或在出现异常情况时进行校正。 2.1.3.1溶出度校正片分崩解型和非崩解型两种崩解型为泼尼松片非崩解型为水杨酸片。目前国内仅有非崩解型校正片。 2.1.3.2校正前应先调式所用仪器。 2.1.3.3溶剂磷酸盐缓冲液 PH7.4。配制方法见中国药典2000年版二部附录XV D要求PH值为7.40?0.05临用前脱气。 2.1.3.4对照品溶液的制备取溶出度 乳体中研细精密称取适量约相当于水杨酸10mg置校正用水杨酸片1片精密称定置