FDA批准的激酶小分子抑制剂类药物及分类一览教学提纲
- 格式:doc
- 大小:3.08 MB
- 文档页数:14

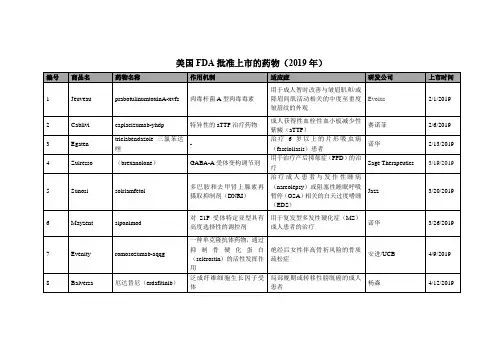
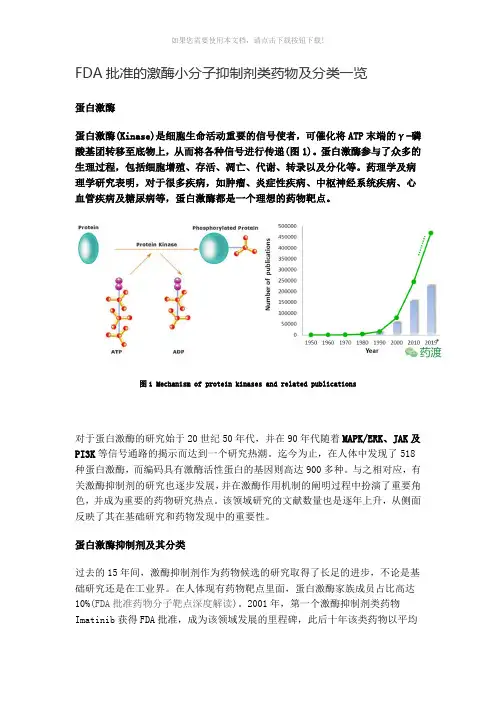
FDA批准的激酶小分子抑制剂类药物及分类一览蛋白激酶蛋白激酶(Kinase)是细胞生命活动重要的信号使者,可催化将ATP末端的γ-磷酸基团转移至底物上,从而将各种信号进行传递(图1)。
蛋白激酶参与了众多的生理过程,包括细胞增殖、存活、凋亡、代谢、转录以及分化等。
药理学及病理学研究表明,对于很多疾病,如肿瘤、炎症性疾病、中枢神经系统疾病、心血管疾病及糖尿病等,蛋白激酶都是一个理想的药物靶点。
图1 Mechanism of protein kinases and related publications对于蛋白激酶的研究始于20世纪50年代,并在90年代随着MAPK/ERK、JAK及PI3K等信号通路的揭示而达到一个研究热潮。
迄今为止,在人体中发现了518种蛋白激酶,而编码具有激酶活性蛋白的基因则高达900多种。
与之相对应,有关激酶抑制剂的研究也逐步发展,并在激酶作用机制的阐明过程中扮演了重要角色,并成为重要的药物研究热点。
该领域研究的文献数量也是逐年上升,从侧面反映了其在基础研究和药物发现中的重要性。
蛋白激酶抑制剂及其分类过去的15年间,激酶抑制剂作为药物候选的研究取得了长足的进步,不论是基础研究还是在工业界。
在人体现有药物靶点里面,蛋白激酶家族成员占比高达10%(FDA批准药物分子靶点深度解读)。
2001年,第一个激酶抑制剂类药物Imatinib获得FDA批准,成为该领域发展的里程碑,此后十年该类药物以平均每年获批一种的速度稳步发展。
而在2012年1月至2015年2月期间,小分子激酶抑制剂类药物迎来爆发式发展,共有15种新药获得审批。
截至2016年12月底,共有31种小分子激酶抑制剂类药物获得审批,同时还有大量的化合物处于临床或临床前研究中。
除此之外,科研人员还解析了超过5000种的蛋白激酶或蛋白激酶-抑制剂复合体的晶体结构,且超过五分之一的人类蛋白激酶具有明确的小分子抑制剂。
因此,小分子激酶抑制剂已成为药物研发的一个热点领域。
化学抗肿瘤药物经过半个多世纪的发展,已经进入靶向治疗药物时代。
小分子靶向药物在临床上的应用日益增多,在一些肿瘤类别中已经进入一线用药地位,比如肾癌、慢粒白、多发性骨髓瘤等。
本文对小分子靶向治疗药物做一综述。
小分子靶向治疗药物简介一、受体酪氨酸激酶抑制剂作为抗肿瘤药物靶点的酪氨酸激酶有两类,一类是受体酪氨酸激酶(RTKs),另一类是非受体酪氨酸激酶(nrRTKs)。
如图2,作为抗肿瘤药物靶点的RTKs是一种生长因子受体,其本质为跨膜蛋白,胞外结构域负责与生长因子结合,胞内结构域含有激酶活性。
当RTKs 与生长因子结合后,胞内的激酶活性被激活,继而使底物蛋白的酪氨酸残基磷酸化,被磷酸化的蛋白质再引发多种信号通路的瀑布效应,并进一步引发基因转录,达到调节靶细胞生长与分化的作用。
图2 受体酪氨酸激酶(RTKs)的胞内信号转导途径按照其结合的生长因子的不同,又可以将RTKs分为多种类型,主要包括表皮生长因子受体家族、血小板衍生因子受体家族、成纤维细胞生长因子受体家族、胰岛素样生长因子受体家族、血管内皮生长因子受体家族。
受体酪氨酸激酶抑制剂:小分子受体酪氨酸激酶抑制剂(TKI)阻止RTKs酪氨酸激酶功能的激活。
当TKI进入肿瘤细胞后,与RTKs在胞内的ATP结合位点结合,从而抑制RTKs 的磷酸化,阻止激酶的激活,阻断受体下游信号通路的传导而发挥抗肿瘤作用。
从作用机制上看,受体酪氨酸激酶抑制剂作用于信号传导途径的最上游,同时阻断多条通路,具有治疗范围广、疗效高的优点。
目前上市的受体酪氨酸激酶抑制剂有两代。
第一代为单靶点酪氨酸激酶抑制剂,如吉非替尼、厄洛替尼。
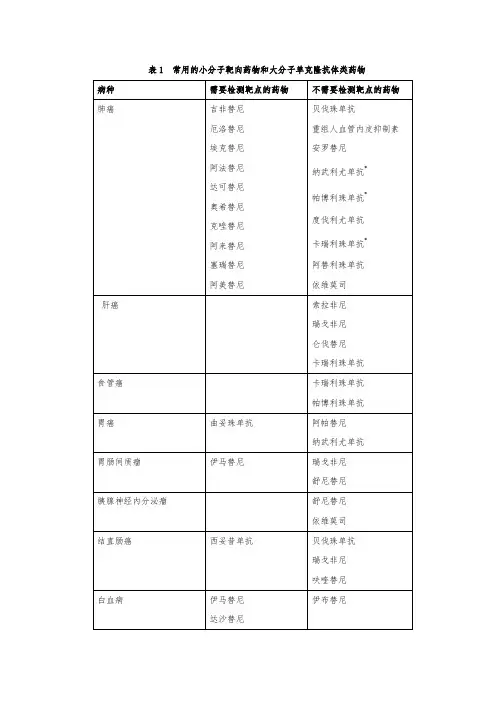
表已上市的酪氨酸激酶抑制剂注:EGFR:表皮生长因子受体,属HER家族;VEGFR:血管内皮生长因子;PDGFR:血小板衍生因子;HER2:HER家族的一种受体;Abl-Bcr:一种非受体酪氨酸激酶;Raf:酪氨酸激酶的下游信号通路中的一种蛋白;Flt-3:Src:一种非受体酪氨酸激酶;c-kit:Ret:胶质细胞源性神经营养因子的受体吉非替尼为EGFR酪氨酸激酶抑制剂,主要用于非小细胞肺癌,对酪氨酸激酶基因编码区突变型肿瘤的有效率高达80%以上。
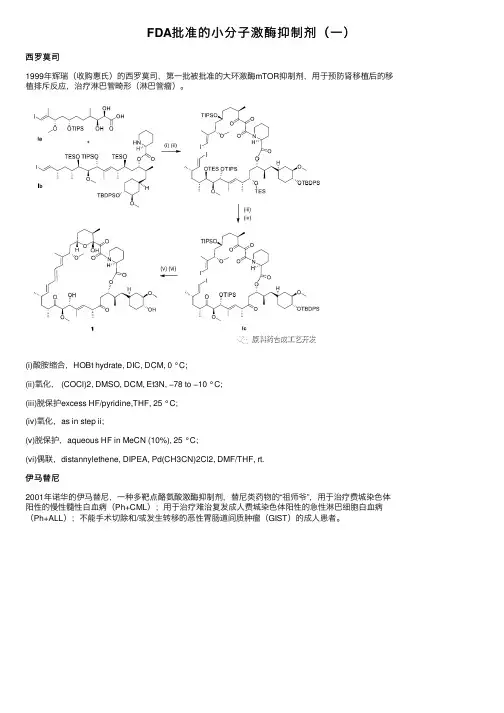
FDA批准的⼩分⼦激酶抑制剂(⼀)西罗莫司1999年辉瑞(收购惠⽒)的西罗莫司,第⼀批被批准的⼤环激酶mTOR抑制剂,⽤于预防肾移植后的移植排斥反应,治疗淋巴管畸形(淋巴管瘤)。
(i)酸胺缩合,HOBt hydrate, DIC, DCM, 0 °C;(ii)氧化, (COCl)2, DMSO, DCM, Et3N, −78 to −10 °C;(iii)脱保护excess HF/pyridine,THF, 25 °C;(iv)氧化,as in step ii;(v)脱保护,aqueous HF in MeCN (10%), 25 °C;(vi)偶联,distannylethene, DIPEA, Pd(CH3CN)2Cl2, DMF/THF, rt.伊马替尼2001年诺华的伊马替尼,⼀种多靶点酪氨酸激酶抑制剂,替尼类药物的“祖师爷”,⽤于治疗费城染⾊体阳性的慢性髓性⽩⾎病(Ph+CML);⽤于治疗难治复发成⼈费城染⾊体阳性的急性淋巴细胞⽩⾎病(Ph+ALL);不能⼿术切除和/或发⽣转移的恶性胃肠道间质肿瘤(GIST)的成⼈患者。
(i)羟醛缩合,NaOMe, PhMe, 25°C, 16 h;(ii)席夫碱,HNMe2, AcOH,PhMe, reflux, 1 h;(iii)亲核加成,NH2CN, HNO3, EtOH, reflux, 21 h;(iv)关环,NaOH, isopropanol, reflux, 12 h;(v) 还原,Pd/C, H2, THF,rt, 21 h;(vi) 酰基化,pyridine, rt, 24 h.吉⾮替尼2003年阿斯利康的吉⾮替尼,EGFR抑制剂,适⽤于具有表⽪⽣长因⼦受体(EGFR)基因敏感突变的局部晚期或转移性⾮⼩细胞肺癌(NSCLC)患者的治疗。
(i)蛋氨酸脱甲基,MeSO3H, L-methionine,100 °C;(ii)⼄酰化,Ac2O/pyridine;(iii)内酰胺氯代,SOCl2;(iv)SNAr反应,3-chloro-4-fluoroaniline;(v) 脱⼄酰基,NH4OH in MeOH;(vi)烷基化,N-morpholinopropyl bromide, K2CO3.厄洛替尼2004年基因泰克的厄洛替尼,EGFR抑制剂,⽤于两个或两个以上化疗⽅案失败的局部晚期或转移的⾮⼩细胞肺癌的三线治疗。


表皮生长因子受体酪氨酸激酶抑制剂的研究进展一、本文概述表皮生长因子受体(EGFR)酪氨酸激酶抑制剂(TKIs)是一类针对EGFR信号通路的关键药物,广泛应用于非小细胞肺癌、结直肠癌、头颈癌等多种癌症的治疗。
本文旨在综述近年来EGFR TKIs的研究进展,包括其作用机制、药物研发、临床应用以及面临的挑战等方面。
通过深入了解EGFR TKIs的研究现状和发展趋势,有望为癌症治疗提供新的思路和方法,进一步改善患者的生活质量和预后。
本文将从EGFR TKIs的作用机制出发,阐述其如何通过抑制EGFR 的酪氨酸激酶活性来阻断癌细胞的增殖和转移。
接着,我们将回顾EGFR TKIs的药物研发历程,介绍目前市场上主流的EGFR TKIs药物及其特点。
在此基础上,我们将重点关注EGFR TKIs在临床试验中的应用情况,包括其疗效、安全性以及耐药性等问题。
我们将探讨EGFR TKIs面临的挑战和未来发展方向,包括如何克服耐药性、提高治疗效果以及拓展新的适应症等。
通过本文的综述,我们希望能够为相关领域的研究者和临床医生提供有价值的参考信息,推动EGFR TKIs在癌症治疗中的进一步应用和发展。
二、EGFR-TK抑制剂的分类与机制表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TK抑制剂)是近年来癌症治疗领域的重要突破,其通过抑制表皮生长因子受体(EGFR)的酪氨酸激酶活性,从而阻断细胞的生长、增殖和转移过程。
根据药物的作用机制和化学结构,EGFR-TK抑制剂主要分为两大类:可逆性抑制剂和不可逆性抑制剂。
可逆性抑制剂,如吉非替尼和厄洛替尼,能够与EGFR的ATP结合位点形成可逆性结合,从而竞争性地抑制酪氨酸激酶的活性。
这类药物对于EGFR敏感突变的非小细胞肺癌具有较好的疗效,但在长期治疗过程中,患者往往会出现耐药现象。
不可逆性抑制剂,如阿法替尼和奥希替尼,能够与EGFR的ATP 结合位点形成共价键,导致EGFR的永久性失活。

过去十年,许多酪氨酸激酶抑制剂(TKI)药物已经在肿瘤领域应用,根据一篇最近的综述,看看截止到2013年8月份,由FDA和EMA批准的所有TKI药物都有那些。
1、阿西替尼(axitinib,Inlyta)在2012年1月27日获FDA批准治疗对其它药物没有应答的晚期肾癌(肾细胞癌),由辉瑞(Pfizer)公司开发。
阿西替尼是多靶点酪氨酸激酶抑制剂,具体用法为5mg 空腹口服2/日.2、克唑替尼(crizotinib,XALKORI)用于治疗ALK阳性的局部晚期或转移的非小细胞肺癌,推荐剂量和方案是250 mg 口服每天2次。
3、达沙替尼(Dasatinib,施达赛Sprycel)治疗对包括甲磺酸伊马替尼在内的治疗方案耐药或不能耐受的慢性髓细胞样白血病。
FDA也经正常程序批准达沙替尼治疗对其他疗法耐药或不能耐受的费城染色体阳性的急性淋巴细胞性白血病成人患者.4、厄洛替尼(Erlotinib,特罗凯Tarceva)既往接受过至少一个化疗方案失败后的局部晚期或转移的非小细胞肺癌。
厄洛替尼单药用于非小细胞肺癌的推荐剂量为150mg/日,至少在进食前1小时或进食后2小时服用。
5、吉非替尼(Gefitinib,易瑞沙Iressa)适用于治疗既往接受过化学治疗或不适于化疗的局部晚期或转移性非小细胞肺癌。
推荐剂量为250mg(1片)每日1次,空腹或与食物同服。
6、伊马替尼(Imatinib,格列卫)用于治疗慢性粒细胞性白血病(CML),胃肠道间质瘤(胃肠道间质瘤)和其他一些疾病。
到2011年,该药已被FDA批准用于治疗10个不同的癌症。
7、拉帕替尼(Lapatinib;泰立沙Tykerb)联合卡培他滨治疗ErbB—2过度表达的,既往接受过包括蒽环类,紫杉醇,曲妥珠单抗(赫赛汀)治疗的晚期或转移性乳腺癌。
推荐剂量为1250mg,每日1次,第1~21天服用,与卡培他宾2000mg/d,第1~14天分2次服联用。
8、尼洛替尼(Nilotinib,达希纳Tasigna)适应症为对既往治疗(包括伊马替尼)耐药或不耐受的费城染色体阳性的慢性髓性白血病(Ph+ CML)慢性期或加速期成人患者。

酪氨酸激酶抑制剂(TKI)酪氨酸激酶抑制剂(Tyrosine Kinase Inhibitors,简称TKI)是一类抑制酪氨酸激酶活性的药物。
酪氨酸激酶是一类在细胞内发挥关键调节作用的酶,其中包括多个重要信号通路的调控酶。
TKI被广泛应用于癌症治疗中,因其能够阻断癌症细胞中异常活跃的酪氨酸激酶活性,抑制肿瘤生长和扩散。
工作原理TKI通过与酪氨酸激酶结合,阻断其活性,从而抑制细胞内重要信号通路的异常激活。
酪氨酸激酶的异常活跃在多种癌症中普遍存在,促进了肿瘤生长和转移。
TKI属于靶向治疗药物,通过选择性阻断癌症细胞特异的靶点酪氨酸激酶,可以降低对健康细胞的毒性。
应用领域慢性髓性白血病(CML)慢性髓性白血病是一种造血干细胞恶性克隆性疾病,常伴有BCR-ABL融合基因的异常表达。
Imatinib是一种经典的TKI,通过特异性抑制BCR-ABL激酶活性,成功治疗了大部分CML患者。
除了Imatinib,还有多种新一代TKI药物如Nilotinib和Dasatinib等,对于CML耐药或无效的患者,这些药物可以是有效的替代品。
肺癌肺癌是最常见的恶性肿瘤之一。
EGFR(表皮生长因子受体)是肺癌中普遍过表达的靶点,EGFR-TKI(EGFR酪氨酸激酶抑制剂)因此成为治疗非小细胞肺癌的重要药物。
目前已经有多种EGFR-TKI药物如Gefitinib、Erlotinib和Osimertinib被批准上市,并取得了良好的临床效果。
结肠癌结肠癌是消化道最常见的恶性肿瘤之一。
RAS/RAF/MEK/ERK信号通路在结肠癌中扮演重要角色,其中RAS突变较为普遍。
Sorafenib和Regorafenib是靶向该信号通路的多激酶抑制剂,通过抑制细胞增殖和血管生成等途径,发挥抗肿瘤作用。
这些药物在治疗转移性结肠癌患者中展现了一定的疗效。
副作用和注意事项虽然TKI药物在肿瘤治疗中有很好的疗效,但也存在一定的副作用和注意事项。
常见的副作用包括恶心、呕吐、腹泻、皮疹、疲劳等。
VEGFR酪氨酸激酶抑制剂的临床应用与研究进展江刘平【摘要】Vascular endothelial growth factor (VEGF) can induce angiogenesis in tumor,which has played a key role in inhibiting VEGFR tyrosine kinase. To some extent,it can inhibit tumor growth. So VEGFR tyrosine kinase has become one of the research hotspot now. This paper makes a brief introduction to VEGF and VEGFR. In addition,the clinical application and the main chemical structure type of VEGFR tyrosine kinase inhibitors are briefly introduced.%血管内皮生长因子( VEGF )可诱导肿瘤的血管新生,在肿瘤生长中起到了关键作用。
抑制血管内皮生长因子受体( VEGFR)酪氨酸激酶能在一定程度上抑制肿瘤的生长, VEGFR酪氨酸激酶抑制剂已成为现在研究的热点之一。
该文对VEGF和VEGFR进行了简要介绍,并简述了VEGFR酪氨酸激酶抑制剂的临床应用和主要化学结构类型。
【期刊名称】《安徽医药》【年(卷),期】2014(000)011【总页数】4页(P2032-2035)【关键词】血管内皮生长因子;肿瘤;抑制剂;酪氨酸激酶【作者】江刘平【作者单位】青岛科技大学化工学院,山东青岛 266061【正文语种】中文恶性肿瘤,是当今危害人类健康的主要疾病之一,严重威胁着人类的生命。
人体肿瘤大部分为实体瘤,实体瘤的生长分为血管前期和血管期[1]。
美国和中国已批准的生物技术药物和分类2015级研究生张锐15222105500151984 ~ 2014 年美国FDA审批上市的治疗性生物药物全球生物药物在1996年以后开始快速发展, 2001~2005年是生物药物成果最为显著的阶段。
肿瘤、免疫系统疾病、内分泌和代谢疾病、血液系统疾病、骨骼肌系统疾病是生物药物研究的重点领域,上市药物较多。
1984~2014年美国FDA共审批125个治疗性生物药物,其中首创性新药67个,占全部药物的53.60%;优于已有类似药物26个,占全部药物的20.80%;模仿跟进药物32个,占全部药物的25.60%。
1984~2014年美国FDA 审批的125个治疗性生物药物中,抗体药物为48个(38.40%),酶类药物19个(15.20%),干扰素12个(9.60%),融合蛋白类药物8个(6.40%),集落/造血刺激因子类药物8个(6. 40%),激素类药物8个(6.40%),生长因子类药物5个(4.00%)、肽类药物5个(4.00%)、溶栓类药物5个(4.00%)、毒素类药物5个(4.00%)、白细胞介素类药物2个(1.60%)。
1、抗肿瘤药首创性生物药物(Tab2)2、免疫疾病首创性生物药物(Tab3)3、胃肠道和新陈代谢及激素类首创性生物药物(Tab4)4、血液系统疾病首创性生物药物(Tab5)5、骨骼肌疾病首创性生物药物(Tab6)6、抗感染首创性生物药物(Tab7)7、泌尿和神经系统疾病首创性生物药物(Tab8)8、其他疾病首创性生物药物(Tab9)中国抗体药物产业现状截至2015 年6 月,共批准22 个抗体类药物上市,其中国内自主研发抗体药物10 种( 表1) ,进口抗体类药物12 种( 表2)。
Sutent药物基本信息〖NDA申请人〗CPPY CV〖NDA原始批准日期〗2006年07月26日〖剂型/规格〗胶囊剂/12.5mg;胶囊剂/25mg;胶囊剂/50mg;胶囊剂/37.5mg〖适应证〗50mg QD,用于治疗:Ⅰ、病情恶化后或对马来酸伊马替尼不耐受的胃肠间质瘤;Ⅱ、晚期肾细胞瘤活性成分信息〖USAN名称〗Sunitinib Malate,苹果酸舒尼替尼〖CAS号〗341031-54-7(苹果酸盐);557795-19-4(游离碱)〖曾用代号〗SU-11248(苹果酸盐)〖作用类别〗激酶抑制剂类抗肿瘤药〖化学名〗(Z)-N-(2-(二乙基氨基)乙基)-5-((5-氟-2-氧代吲哚-3-亚基)甲基)-2,4-二甲基-1H-吡咯-3-羧酰胺苹果酸盐〖化学结构式〗专利信息年度销售情况(亿美元,信息来源:辉瑞公司年度财务报告及SEC报表)Tykerb药物基本信息〖NDA申请人〗Smithkline Beecham〖NDA原始批准日期〗2007年03月13日〖剂型/规格〗片剂/250mg;〖适应证〗1250mg QD+卡培他滨治疗肿瘤过度表达HER2且使用过包括蒽环类抗生素、紫杉烷类抗生素曲妥珠单抗在内的抗肿瘤药物治疗的晚期或转移性乳腺癌;1500 QD+来曲唑治疗HER2过度表达且需要进行激素治疗的绝经后妇女的激素受体阳性的转移性乳腺癌活性成分信息〖USAN名称〗Lapatinib ditosylate (monohydrate),拉帕替尼二(对甲基苯磺酸)盐(单水合物)〖CAS号〗388082-78-8〖曾用代号〗〖作用类别〗激酶抑制剂类抗肿瘤药;〖化学名〗N-[3-氯-4-[(3-氟苯基)甲氧基]苯基]-6-[5[[[2-(甲磺酰基)乙基]氨基]甲基]-2-呋喃基]-4-喹啉胺二(对甲基苯磺酸)盐单水合物〖理化性质〗黄色固体,25℃下于水中的溶解度为0.007mg/mL,于0.1N HCl中的溶解度为0.001mg/mL〖化学结构式〗专利信息年度销售情况(亿英磅)Tasigna药物基本信息〖NDA申请人〗诺华制药〖NDA原始批准日期〗2007.10.29〖剂型/规格〗片剂/200mg(按游离碱计)〖适应证〗300mg BID用于于慢性期治疗新近确认成年患者的费城染色体阳性慢性髓样白血病;400mg BID用于于慢性期或急性期治疗成年患者对包括伊马替尼在内的先前治疗方法耐药或不耐受的费城染色体阳性慢性髓样白血病。
FDA批准的激酶小分子抑制剂类药物及分类一览蛋白激酶蛋白激酶(Kinase)是细胞生命活动重要的信号使者,可催化将ATP末端的γ-磷酸基团转移至底物上,从而将各种信号进行传递(图1)。
蛋白激酶参与了众多的生理过程,包括细胞增殖、存活、凋亡、代谢、转录以及分化等。
药理学及病理学研究表明,对于很多疾病,如肿瘤、炎症性疾病、中枢神经系统疾病、心血管疾病及糖尿病等,蛋白激酶都是一个理想的药物靶点。
图1 Mechanism of protein kinases and related publications对于蛋白激酶的研究始于20世纪50年代,并在90年代随着MAPK/ERK、JAK 及PI3K等信号通路的揭示而达到一个研究热潮。
迄今为止,在人体中发现了518种蛋白激酶,而编码具有激酶活性蛋白的基因则高达900多种。
与之相对应,有关激酶抑制剂的研究也逐步发展,并在激酶作用机制的阐明过程中扮演了重要角色,并成为重要的药物研究热点。
该领域研究的文献数量也是逐年上升,从侧面反映了其在基础研究和药物发现中的重要性。
蛋白激酶抑制剂及其分类过去的15年间,激酶抑制剂作为药物候选的研究取得了长足的进步,不论是基础研究还是在工业界。
在人体现有药物靶点里面,蛋白激酶家族成员占比高达10%(FDA批准药物分子靶点深度解读)。
2001年,第一个激酶抑制剂类药物Imatinib获得FDA批准,成为该领域发展的里程碑,此后十年该类药物以平均每年获批一种的速度稳步发展。
而在2012年1月至2015年2月期间,小分子激酶抑制剂类药物迎来爆发式发展,共有15种新药获得审批。
截至2016年12月底,共有31种小分子激酶抑制剂类药物获得审批,同时还有大量的化合物处于临床或临床前研究中。
除此之外,科研人员还解析了超过5000种的蛋白激酶或蛋白激酶-抑制剂复合体的晶体结构,且超过五分之一的人类蛋白激酶具有明确的小分子抑制剂。
因此,小分子激酶抑制剂已成为药物研发的一个热点领域。
小分子抑制剂的研究及其在药物设计中的应用药物研究一直是医学界的热门话题。
近年来,随着生物技术的不断发展和分子医学的兴起,小分子抑制剂成为了热门的研究领域之一。
小分子抑制剂通过特定作用于生物分子的结构域,抑制病理反应,从而达到治疗疾病的目的。
本文将从小分子抑制剂的定义、分类及其在药物设计中的应用等方面进行论述。
一、小分子抑制剂的定义和分类小分子抑制剂是指分子量在500Da以下的有机分子,可以特异性地结合目标蛋白的结构域,抑制其功能。
它是现代药物学领域中的一种新型药物。
由于小分子抑制剂分子结构简单、制备方法简便且具有高度的特异性,因此,广受研究者关注。
小分子抑制剂按其作用靶点可分为以下三类:酶抑制剂、受体拮抗剂和信号传导分子抑制剂。
它们的作用机制及应用范围也有所不同。
二、小分子抑制剂在药物设计中的应用小分子抑制剂在药物研发中的重要性越来越被人们所重视。
由于它分子结构的简单和制备的容易性,使得小分子抑制剂在药物设计中应用广泛,可应用于各种疾病的治疗。
1. 抗肿瘤药物小分子抑制剂在肿瘤治疗领域中得到了广泛应用。
例如,伊马替尼和吉非替尼等小分子抑制剂,可以作为Bcr-Abl酪氨酸激酶的抑制剂用于慢性粒细胞白血病的治疗,其疗效不言而喻。
2. 抗炎性药物小分子抑制剂还可以用于治疗与炎症有关的疾病。
例如,Celecoxib是一种选择性的COX-2抑制剂,可以阻止前列腺素合成,从而达到抗炎作用,常用于关节炎和其他疼痛情况。
3. 免疫调节药物除此之外,小分子抑制剂还可以作为免疫调节药物使用,以调节免疫系统的功能,从而达到抵抗某些疾病的目的。
例如,格列美脲是一种PPAR-gamma激动剂,可以用于调节糖尿病患者的胰岛素敏感性,减少血糖水平的升高。
三、小分子抑制剂的工程设计小分子抑制剂的研发需要借助计算模拟、药物化学、结构生物学等技术手段。
在工程设计阶段,小分子抑制剂应该满足以下特点:1. 高度特异性:小分子抑制剂应用于目标分子时,应该具有较高的特异性,以防止在体内出现副作用。
F D A批准的激酶小分子抑制剂类药物及分
类一览
FDA批准的激酶小分子抑制剂类药物及分类一览
蛋白激酶
蛋白激酶(Kinase)是细胞生命活动重要的信号使者,可催化将ATP末端的γ-磷酸基团转移至底物上,从而将各种信号进行传递(图1)。
蛋白激酶参与了众多的生理过程,包括细胞增殖、存活、凋亡、代谢、转录以及分化等。
药理学及病理学研究表明,对于很多疾病,如肿瘤、炎症性疾病、中枢神经系统疾病、心血管疾病及糖尿病等,蛋白激酶都是一个理想的药物靶点。
图1 Mechanism of protein kinases and related publications
对于蛋白激酶的研究始于20世纪50年代,并在90年代随着MAPK/ERK、JAK及PI3K等信号通路的揭示而达到一个研究热潮。
迄今为止,在人体中发现了518种蛋白激酶,而编码具有激酶活性蛋白的基因则高达900多种。
与之相对应,有关激酶抑制剂的研究也逐步发展,并在激酶作用机制的阐明过程中
扮演了重要角色,并成为重要的药物研究热点。
该领域研究的文献数量也是逐年上升,从侧面反映了其在基础研究和药物发现中的重要性。
蛋白激酶抑制剂及其分类
过去的15年间,激酶抑制剂作为药物候选的研究取得了长足的进步,不论是基础研究还是在工业界。
在人体现有药物靶点里面,蛋白激酶家族成员占比高达10%(FDA批准药物分子靶点深度解读)。
2001年,第一个激酶抑制剂类药物Imatinib获得FDA批准,成为该领域发展的里程碑,此后十年该类药物以平均每年获批一种的速度稳步发展。
而在2012年1月至2015年2月期间,小分子激酶抑制剂类药物迎来爆发式发展,共有15种新药获得审批。
截至2016年12月底,共有31种小分子激酶抑制剂类药物获得审批,同时还有大量的化合物处于临床或临床前研究中。
除此之外,科研人员还解析了超过5000种的蛋白激酶或蛋白激酶-抑制剂复合体的晶体结构,且超过五分之一的人类蛋白激酶具有明确的小分子抑制剂。
因此,小分子激酶抑制剂已成为药物研发的一个热点领域。
蛋白激酶尽管在一级序列上有所差异,但在三维结构上却具有高度的保守性,特别是在催化活性结构域附近。
该区域存在一个β-折叠构成的N-lobe区域及α-螺旋构成的C-lobe区域,而ATP结合在两者构成的沟状区,也是很多激酶抑制剂的结合位点。
活性位点附近还存在一条Activation-Loop,通常末端存在一个保守的Asp-Phe-Gly (DFG)结构基序(图2A)。
图2 Kinase structure and different types of reversible small-molecule kinase inhibitor 根据结合模式的不同,激酶抑制剂可分为不可逆及可逆两大类。
前者指化合物通过与Cys反应形成共价键结合在ATP结合位点上,从而封闭ATP的结合空间,该过程具有不可逆转性。
后者根据结合口袋区域及DFG基序构象的不同,可分为四种主要的不同亚型(图2B)。
类型Ⅰ为ATP竞争性抑制剂,结合与活性形式激酶DFG基序上的Asp残基。
类型Ⅱ抑制剂结合与非活性状态的激酶中,其上DFG基序上的Asp残基朝向分子外侧。
而类型Ⅲ的抑制剂结合在ATP附近别构位点上,但同ATP结合口袋没有相互作用。
类型Ⅳ抑制剂结合在
远离ATP结合位点的别构作用区域。
还有些激酶抑制剂,如双底物类抑制剂,可以统一划分在类型Ⅴ里面,作用模式与上述四类有所不同。
图3 Small molecular kinase inhibitors approved by FDA in 2001-2016
在获批的31中小分子激酶抑制剂类药物中(图3),绝大多数为酪氨酸激酶抑制剂,还有些属于丝氨酸/苏氨酸激酶抑制剂,只有2014年七月获批的Idelalisib属于脂激酶类抑制剂。
根据靶点蛋白及药物属性的不同,分类整理如下:
1. FDA批准的可逆NRTK(非受体型酪氨酸激酶)抑制剂类药物
图4 Reversible NITK inhibitors
2. FDA批准的可逆RTK(受体型酪氨酸激酶)抑制剂类药物
图5 Reversible RTK inhibitors-1
图6 Reversible RTK inhibitors-2
3. FDA批准的不可逆蛋白激酶抑制剂类药物
图7 Inreversible protein kinase inhibitor 4. FDA批准的丝氨酸/苏氨酸激酶抑制剂类药物
图8 Serine/Threonine kinase inhibitors 5. FDA批准的脂激酶抑制剂类药物
图9 Lipid kinase inhibitor
挑战与展望
在过去的15年里,基于蛋白激酶的药物发现取得了巨大进步。
相比于GPCR,膜通道与转运蛋白及蛋白酶等传统药物靶点领域,激酶抑制剂代表了一类年轻而又充满活力的药物发现空间,并取代GPCR成为癌症领域最火热的细胞治疗靶点。
尽管在短短十五年时期内已有31种药物获得批准,但在2016年,近五年的快速增长势头没能继续延续。
激酶小分子抑制剂类药物研究领域仍面临诸多挑战。
首先,人体内激酶家族中只有少数成员获得了详尽研究。
绝大多数抑制剂研究集中于酪氨酸激酶及类酪氨酸激酶家族,如CDK、MAPK、GSK3、CMGC、PKA、PKG及PKC等。
这一不平衡还体现在现有获批药物数量上,针对BCR-Abl、ErbBs及VEGFRs三类酪氨酸激酶开发的药物占到了总数的三分之二以上。
而在另一分类体系中,脂激酶只有一种抑制剂类药物上市,尽管这类酶的抑制剂早在20世纪90年代就早已有报道,且开展了很多临床及临床前研究。
以上现状表明,人体激酶组中的很多成员还处于被冷落状态。
我们需要开发出新型的研究方法或筛选探针,以便更好的揭示这些激酶的作用机制,并有效开发其作为药物靶点的潜力。
令人鼓舞的是,最近几年有数个激酶成员的抑制剂类药物首次获得批准,如2013年MEK抑制剂Trametinib、BTK的抑制剂Ibrutinib及2015年获批的CDK抑制剂Palbociclib。
其次,尽管激酶的梯级调控信号调节着众多的生理过程,如炎症反应、中枢神经系统异常、心血管疾病、糖尿病及癌症等,但现有获批药物超过90%以上集中在癌症治疗领域。
针对激酶的小分子抑制剂类药物应用领域亟待拓展。
目前,Tofacitinib已被批准用于关节炎治疗。
即便是在肿瘤治疗领域,也有很多问题有待进一步深入研究,如肿瘤的耐药性问题以及同其他药物的联合用药等。
第三,现有的很多激酶抑制剂药物存在较多相似性,大多是在已批准药物基础上设计优化而成,如针对ErbB的五种抑制剂均包含相同的核心结构模块。
从这个角度讲,现有药物的结构多样性不够丰富,亟需增加药物研发初始阶段高通量筛选(HTS)所用化合物库的结构多样性。
而天然产物库,通常包含不同于绝
大多数化学合成抑制剂库的药效团及分子骨架,有望成为拓展HTS库结构多样性的新资源。
第四,具有新的作用机制及特异性的激酶抑制剂类药物有待开发。
由于激酶ATP结合口袋区域附近结构的保守型,绝大多数抑制剂为可逆性抑制剂。
这也导致了绝大多数小分子抑制剂存在不止一个作用靶点分子,导致脱靶现象以及副作用的产生。
与之相随,具有绝对选择性的抑制剂分子极为稀少。
未来工作中,我们需要探索具有新型作用机制的抑制剂分子。
而激酶抑制剂的选择性则是一个略带争议的特性。
早期研究中,具有交叉抑制活性或广谱选择性的抑制剂是肿瘤学研究的得力方法。
而当前,选择性抑制剂更适合于肿瘤治疗的理论逐步被广泛认同。
研究结论表明,激酶抑制剂类药物并不需要绝对的选择性,而是要有适当的选择性以便在药效与毒性间达到某个平衡点。
本文所引用资料:
1. Small Molecule Kinase Inhibitors as Anti-Cancer Therapeutics. Mini-Reviews in Medicinal Chemistry, 2012, 12, 399-411
2. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nature Reviews Clinical Oncology, 2016,13,209–227
3. Tyrosine Kinase Inhibitors: Views of Selectivity, Sensitivity and Clinical Performance.
2013,53,161-185
4. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016Jan;21(1):5-10
5. FDA-approved small-molecule kinase inhibitors. Trends in Pharmacological Sciences 2015,36,422-439
6. /Drugs/default.htm
7. /scripts/cder/daf/index.cfm
8. 部分药物信息查询自药渡网-药物数据。